
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1Womens College Of Pharmacy, Peth Vadgaon, Maharashtra, India
2Ashokrao mane institute of pharmacy Ambap, Maharashtra, India
Thalassemia represents a major global health burden, particularly in South Asia, the Mediterranean, and the Middle East, where carrier rates are high. Despite improvements in diagnosis and supportive care, significant challenges remain in managing chronic anemia, iron overload, and multi-organ complications. A key clinical dilemma is the management of non-transfusion-dependent thalassemia (NTDT), which is often underrecognized despite its associated morbidity.This review synthesizes current evidence on the pathophysiology, diagnosis, and management of thalassemia, with emphasis on both transfusion-dependent and non-transfusion-dependent forms. Advances in diagnostic approaches, including molecular and prenatal testing, have improved early detection. Established treatments such as regular blood transfusion and iron chelation therapy remain central to care, while emerging strategies—including fetal hemoglobin inducers and gene therapy—offer promising disease-modifying potential.The findings highlight the importance of individualized, risk-based management to prevent long-term complications involving cardiac, hepatic, and endocrine systems. Improved screening, early diagnosis, and continued research into novel therapies are essential to reduce disease burden and enhance patient outcomes.
Thalassemia is an inherited disorder which means they are passed from parents to
their children [1]. Thalassemia is the most common genetic blood disease in the world and varies in different population group in the world [2]. World Health Organization (WHO) estimates that at least 6.5% of the world populations are carries of different inherited disorders of hemoglobin [3]. Thalassemia (also known as Mediterranean anemia, Cooley’s anemia, Beta-thalassemia or Alpha-thalassemia) is an inherited blood disorder affected by an abnormal form of hemoglobin blood disorder is the most common inherited single gene disorder in the world. This specific type of blood disease results in excessive destruction of red blood cells which in turn leads to anemia [4]. For a better understanding of this disease one must know the importance of hemoglobin [5]. In vertebrates, hemoglobin is the iron-containing oxygen-transport protein that is found in red blood cells which carries oxygen from the lungs to the rest of the body and then brings the carbon dioxide back to the lungs to be dispensed. People who have thalassemia produce fewer healthy hemoglobin proteins, and their bone marrow produces fewer healthy red blood cells. With too few normal red blood cells, not enough hemoglobin is available to help carry oxygen to the body [1].
DEFINATION:-
The thalassemias are hereditary disorders characterized by a decrease in the synthesis of globin chains (alpha or beta). Impaired globin chain synthesis causes impaired production of hemoglobin and eventually results in a hypochromic microcytic anemia because of defective hemoglobinization of the red blood cells [6].
CLASSIFICATION OF THALASSEMIA:-
Is due to decreased production of one or more globin chains. The most important types are
those that affect either alpha or beta chain synthesis.
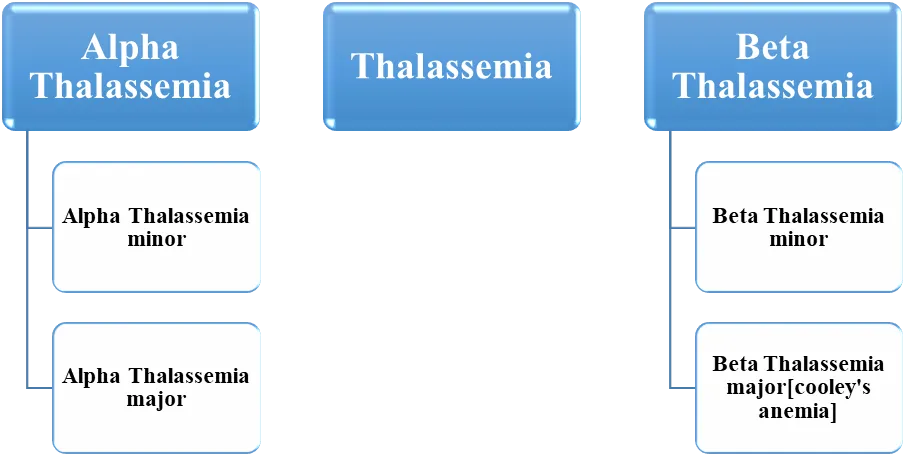
Fig 1. Classification of thalassemia
There are several forms of alpha thalassemia. The most common forms are: Silent carrier
alpha thalassemia: There are two alpha genes located on each chromosome 16 [7]. In the silent carrier state, one of the alpha genes is absent, leaving three of four genes (aa/ao). Patients are hematological healthy, except for occasional low red blood cell indices. Alpha thalassemia trait: This form is usually caused by the deletion of two alpha (a) genes on one chromosome 16 (aa/oo) or the deletion of one gene from each chromosome 16. Alpha thalassemia trait is more common in Southeast Asia, the Indian subcontinent, and some parts of the Middle East. This trait is characterized by mild anemia and low red blood cell indices. Hb H disease: This form results from the deletion or inactivation of three alpha globin genes (oo/ao), resulting also in an excess of beta chains. Patients usually present with severe anemia, splenomegaly, icterus, and abnormal red blood cell indices. Alpha thalassemia major: This condition results from the deletion of all alpha genes on both copies of chromosome 16 (oo/oo), leading to the severe form of homozygous alpha thalassemia [7].
Beta Thalassemia:-
In contrast to the duplication found in alpha thalassemia, there is only one beta-globin gene
on chromosome 11. There are several forms of beta thalassemia: Silent carrier beta thalassemia or beta thalassemia minor: The mutation that causes this form of thalassemia is very mild. These patients usually have no signs or symptoms, or have some minor changes in number or size of the red blood cell. This is the most common form of beta thalassemia. Beta thalassemia trait or beta thalassemia intermediate: In this condition, the production of beta globin is decreased. Patients have mild anemia, abnormal red blood cell indices, and abnormal hemoglobin electrophoresis results with elevated levels of Hb A2, Hb F, or both. Thalassemia major[cooly’s anemia] In this condition, the patient can’t produce beta globin. To compensate, the marrow produces gamma globin and more alpha globin. This condition is characterized by transfusiondependent anemia, massive splenomegaly, bone deformities, growth retardation, and peculiar facies in untreated individuals [8].
ETIOLOGY:-
Thalassemia is due to decreased production of at least one globin polypeptide chain (beta, alpha, gamma, delta) which results in unbalanced hemoglobin synthesis. Inheritance of thalassemia is autosomal recessive. Beta Thalassemia results from decreased production of beta-polypeptide chains. Heterozygotes are carriers and have asymptomatic mild to moderate microcytic anemia (thalassemia minor). Homozygotes (beta-thalassemia major or Cooley’s anemia) develop severe anemia and bone marrow hyperactivity. Alpha thalassemia is a result of decreased production of alpha globins. Heterozygotes for a single gene defect results in silent alpha thalassemia state. Heterozygotes with defects in two of the four genes result in alpha thalassemia trait, and tend to develop mild to moderate microcytic anemia but with no symptoms. Defects in three of the four genes more severely impair alpha-chain production, resulting in the formation of tetramers of excess beta chains (HbH) or, in infancy, gamma chains (Bart’s Hb). Defects in all four genes are a lethal condition unless given blood transfusions in utero, because hemoglobin that lacks alpha chains does not transport oxygen.
Another thalassemia form is hemoglobin E (Hb E) related. Hb E, one of the most common hemoglobinopathies, is due to missense mutation in codon 26 which belongs to splicing sequence of Beta globin gene. This mutation not only affects structure but also reduces the production rate of beta globin. As consequence, Hb E trait and disease usually present with mild Beta thalassemia phenotype. Co-inheritance of Hb E and thalassemia is commonly found in Southeast Asia, India and Bangladesh [9]. This compound heterozygote state of Hb E/Thalassemia results in a variable phenotype ranging from mild anemia to transfusion dependency [10]. The most significant genotype is Hb E/Beta thalassemia which accounts for 50% of severe beta thalassemia worldwide [11]. Patients with Hb E/Beta thalassemia have a higher risk of pulmonary hypertension or vitamin D deficiency [12].
EPIDEMIOLOGY :-
The alpha thalassemia syndromes are seen primarily in persons from Southeast Asia and China, and less commonly, in African Americans.
Beta thalassemia primarily affects persons of Mediterranean origin (Italian, Greek) and to a lesser extent Chinese, other Asians, and blacks.
PATHOPHYSIOLOGY:-
The basic defect in β-thalassemia is a reduced or absent production of β-globin chains with relative excess of α-chains. Because α- and non-α chains pair with each other at a ratio close to 1:1 to form normal Hb, the excess unmatched α chains accumulate in the cell as an unstable product, leading to cell destruction in the bone marrow and in the extramedullary sites. This process is referred to as ineffective erythropoiesis (IE) and is the hallmark of β-thalassemia [13].
The excess α-chains may, in minor amounts, combine with residual β- (in β+ -thalassemia) and γ-chains (whose synthesis persists usually in small quantity after birth), undergo proteolysis, or in large part become associated with the erythroid precursors with deleterious effects on erythroid maturation and survival. Also excess α-chain precipitation in the red cell membrane causes structural and functional alterations with changes in deformability, stability, and red cell hydration [13]
Alterations of erythroid precursors result in an enhanced rate of apoptosis, which is a programmed cell death. Apoptosis could contribute significantly to ineffective erythropoiesis and occurs primarily at the polychromatophilic erythroblast stage. The ineffective erythropoiesis (IE) and anemia have several consequences producing the clinical picture of the disease. The first response to anemia is an increased production of erythropoietin, causing a marked β-Thalassemia erythroid hyperplasia, which may range between 25 and 30 times normal. Anemia may produce cardiac enlargement and sometimes severe cardiac failure [13]. Increased erythropoietin synthesis may stimulate the formation of extramedullary erythropoietic tissue, primarily in the thorax and paraspinal region. Marrow expansion also results in characteristic deformities of the skull and face, as well as osteopenia [14]. High levels of iron, closely associated with denatured hemoglobin, have been found in the membrane of β-thalassemic red cells [15]. Severe IE, chronic anemia, and hypoxia also cause increased gastrointestinal (GI) tract iron absorption. This is combined with increased iron from the breakdown of RBCs and the increased iron introduced into the circulation by the transfusions necessary to treat thalassemia, plus inadequate excretory pathways lead to progressive deposition of iron in tissues and hemosiderosis occurs [14].
Free iron species, such as labile plasma iron as well as labile iron pool in the RBCs accumulate when transferrin saturation exceeds 70%. These free iron species generate reactive oxygen species with eventual tissue damage, organ dysfunction, and death [14].
ASSOCIATED DISORDER :-
Multiple blood transfusions result in iron overload, which can cause secondary hemosiderosis. Cardiac involvement is a major cause of death in patients with thalassemia. Transfusional hemosiderosis has been classified into 3 stages based on the number of blood units given. The higher the number of packed red blood cells units given, the more advanced the stage. Advanced stage is associated with more severe clinical symptoms and more abnormal cardiac functions. Patients who have received regular blood transfusions sometimes develop liver enlargement due to swelling of the phagocytic and parenchymal cells from the deposition of hemosiderin. Calcified bilirubin stones are common in patients with beta thalassemia major. People with thalassemia major frequently exhibit features of diabetes mellitus. This could be due to defective pancreatic production of insulin, but insulin resistance has also been implicated. Adult patients with thalassemia major are known to have low fertility; this is attributed to the commonly encountered hypogonadotrophic hypogonadism. People with thalassemia, especially with thalassemia intermediate, have higher risk of thromboembolism event as compared to the general population [16]. Children or young adults with thalassemia usually present with osteopenia or osteoporosis despite adequate transfusion and iron chelation therapy. The mechanism for osteopathy in thalassemia patient is complex. Osteoporosis reduces bone strength and increases risk of skeletal abnormalities, fractures, spinal deformities, nerve compression, and growth failure [17].
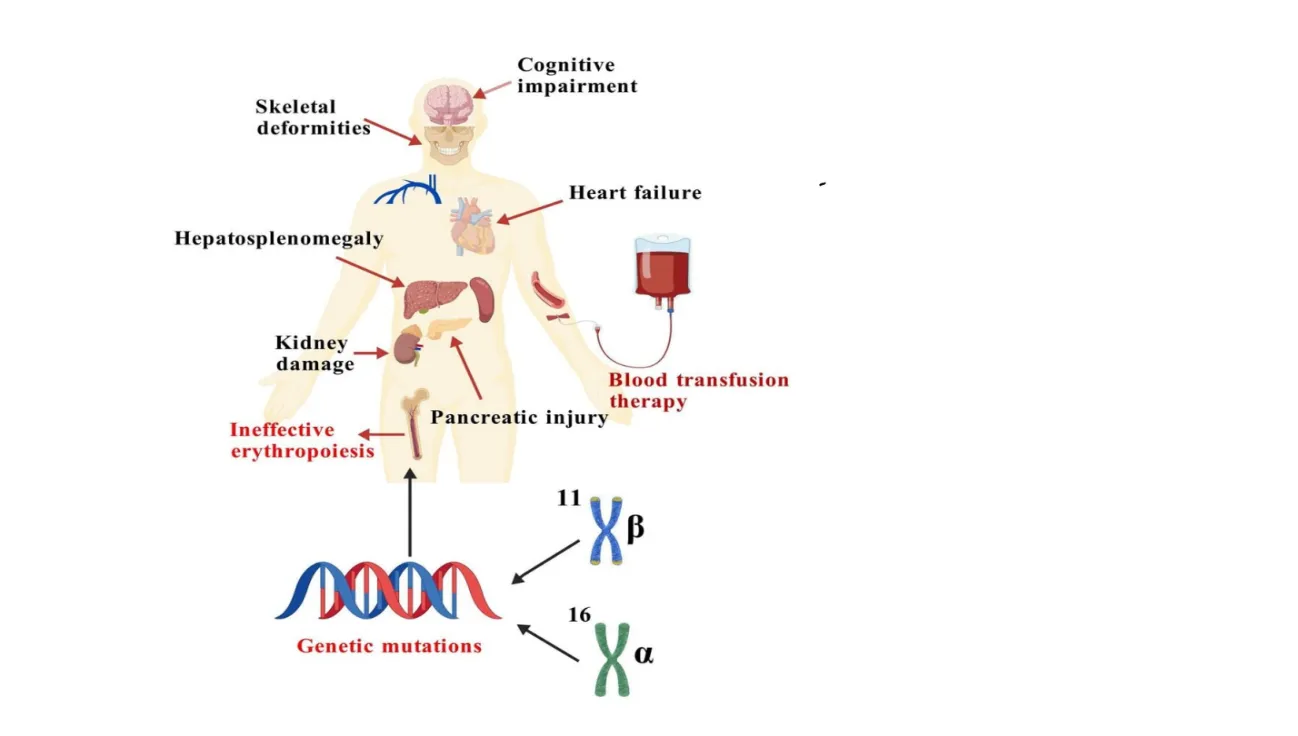
Fig 2. Common clinical manifestations of patients with thalassemia [66]
SYMPTOMS :-
The symptoms of thalassemia are due to the decreased number of healthy red blood cells. Thus, the symptoms you experience depend on how severe your condition is.
Asymptomatic :-You likely won't have any symptoms if you're missing one alpha gene. With two missing alpha genes or one missing beta gene, you may have symptoms of mild anemia, such as fatigue [18].
Mild to moderate symptoms of thalassemia :-Thalassemia intermedia presents with mild to moderate forms of symptoms, such as,
Severe symptoms of thalassemia :-People with hemoglobin H disease or beta thalassemia major have severe forms of thalassemia. Symptoms typically appear within the first two years of life and may include features of severe anemia like dizziness and fast heart rate, along with other health issues such as:
PLANNING A PREGANANCY :-
Pregnancy Planning: Women with severe forms of thalassemia can have successful pregnancies, but you need to consult with your doctor. It's important to consider genetic counseling. It educates you and your partner regarding the nature of thalassemia, its inheritance pattern, and your reproductive options. This also includes discussions on the availability of prenatal and preimplantation genetic testing and the implications of these tests [22,23].
During Pregnancy: Increased monitoring and adjustments to treatment may be necessary, as pregnancy can pose risks such as heart issues for the mother and growth problems for the baby.
Contraception: If pregnancy isn't planned, reliable contraception is recommended [24,25].
SCREENING AND DIAGNOSTIC CRITERIA :-
Diagnosing thalassemia involves a combination of clinical evaluation, laboratory tests, and a genetic analysis to confirm the presence of the disease and determine its severity. Given the heterogeneity of thalassemia phenotypes and the overlap of symptoms with other types of anemia, an accurate diagnosis is essential for appropriate management and treatment planning. Thalassemia screening and diagnostic protocols encompass a range of methods and guidelines. Programs such as the NHS Sickle Cell and Thalassemia Screening Program advocate for antenatal and newborn screening, particularly in areas with high prevalence [26]. Evaluating thalassemia can be challenging due to the overlap of its signs and symptoms with other hematologic disorders. Physicians typically suspect thalassemia based on common features such as anemia, jaundice, hepatosplenomegaly, and growth retardation, which are not exclusive to thalassemia [27,28,29]. Differentiating thalassemia from other conditions requires a comprehensive approach, including genetic testing, hemoglobin electrophoresis, and specific hematologic parameter assessments like the MCV/RBC ratioand HbA2l evels[30]. Further more,studies have shownthatmarkersof erythropoiesis, such as GDF-15 and EPO, are significantly elevated in thalassemia patients, suggesting their potential as diagnostic indicators and therapeutic targets [29]. Thus, a multidisciplinary approach that combines clinical evaluation with advanced diagnostic tools is essential for the accurate diagnosis and management of thalassemia.
1. Laboratory Tests:-
A Complete Blood Count (CBC) is an essential screening tool that aids in detect ing microcytic hypochromic anemia, a common feature of thalassemia and other blood disorders [31]. Parameters such as red blood cell distribution width (RDW) and mean corpuscular volume (MCV) are crucial for distinguishing thalassemia from other types of anemia [32]. The CBC provides valuable insights into the blood’s quantitative and qualitative composition, offering essential information on cell components such as white blood cells, red blood cells, and platelets [33]. Automated hematology analyzers have significantly improved the accuracy and efficiency of CBC results, although challenges like unreliable results due to interfering substances or abnormal cells still require careful consideration [34]. The CBC remains a cornerstone in diagnosing and monitoring various diseases, highlighting its importance in clinical practice [35].
2. Hemoglobin Electrophoresis:-
Hemoglobin electrophoresisisavitaldiagnostictoolthatseparatesdifferenthemoglobin types based on their charge, aiding in the detection of abnormal patterns associated with conditions like thalassemia. Thalassemia typically presents with elevated fetal hemoglobin (HbF) levels and reduced adult hemoglobin (HbA) levels, resulting in distinctive patterns on hemoglobin electrophoresis [36,37]. Moreover, abnormal hemoglobin variants, such as hemoglobin C (Hb C), can affect the accuracy of glycosylated hemoglobin (A1C) tests, underscoring the need to consider these variants in diagnostic evaluations [38]. Elec trophoresis is crucial for the separation and characterization of charged molecules like hemoglobin, providing valuable insights into various hematological disorders and guiding clinical decision-making [39,40].
3. High-Performance Liquid Chromatography (HPLC):-
High-performance liquid chromatography (HPLC) is a method used to separate com pounds or molecules based on their chemical properties. Ion-exchange chromatography is the mosteffective and commonlyusedtechniqueforhemoglobinanalyses. WhileHPLCcan be performed manually, fully automated systems have become available, with some specif ically designed for hemoglobin analyses. In regions with a low prevalence of hemoglobin disorders, systems capable of switching between a glycated hemoglobin analysis for dia betes and a variant hemoglobin analysis for thalassemia may be more practical. HPLC is especially useful for diagnosing the β-thalassemia trait because it allows for an accurate quantification of HbA2. Precise control of analytical conditions, including the column temperature, flow rate, and buffer conditions, is crucial for achieving accurate results [41].
4. Genetic Testing:-
Molecular genetic testing is crucial for confirming thalassemia diagnoses and identify ing specific gene mutations, particularly in the alpha- and beta-globin genes, as highlighted by various studies [42,43]. Techniques such as the polymerase chain reaction (PCR) and DNAsequencing are instrumental in detecting these mutations, providing essential information on disease severity and inheritance patterns [44]. These advanced molecular diagnostic tools have significantly enhanced the sensitivity and specificity of genetic testing, enabling the precise identification of mutation carriers, which is vital for accurate diagnoses and genetic counseling [45]. By leveraging nucleic acid-based approaches, healthcare professionals can confirm the presence of thalassemia, predict disease likelihood, monitor disease progression, and personalize treatment strategies based on genetic variations. Ad ditionally, molecular genetic testing is invaluable for prenatal diagnoses, allowing the early detection of severe hemoglobinopathies and thalassemia in fetuses, thus enabling early intervention and management strategies [41].
TREATMENT:-
Standard therapy includes blood transfusions, folic acid, iron chelating agents, and hematopoietic cell transplant. Treatment choices depend on the patient’s clinical manifestation.
Transfusion therapy:-
The decision to initiate a regular transfusion program in a child newly diagnosed with thalassemia must take into account both laboratory and clinical findings. An overlap of genotype and phenotype expression make the clinical assessment the most important step in distinguishing TM from TI. If the child is growing poorly and has developed facial or other bone abnormalities, and/or when Hb levels are 7 g/dL, regular transfusions will be beneficial [46]. Confounding factors that might aggravate the degree of anemia, including folic acid deficiency and acute febrile illness, blood loss, or coinheritance of glucose-6-phosphate dehydrogenase deficiency, need to be addressed simultaneously with transfusion therapy. If the child is folic acid replete and failing to thrive with no other factors to explain the Hb level of 7 g/dL, a first transfusion is administered. The child is subsequently followed; and when the Hb level falls again to a level of 7 g/dL, a regular monthly transfusion regimen is begun. Before the first transfusion, patients’ RBCs are typed for Rh and ABO antigens. At the same time, cytomegalovirus status should be obtained. Cytomegalovirus-negative blood products are recommended for potential candidates for curative stem cell transplantation (SCT). Parents and first-degree relatives should not be blood donors for these candidates. Hepatitis B vaccination is given before transfusion therapy, as is hepatitis A vaccine when age appropriate [46,47].The risk of transfusion-transmitted infections in thalassemia patients has been greatly reduced since screening for human immunodeficiency virus infections began in 1985 and for hepatitis C in 1991 [47]. However, new agents, such as West Nile Virus and babesiosis, which are not screened for, may contaminate the blood supply from asymptomatic donors [48]. Transfusions of washed, leukocyte-depleted RBCs are recommended for all the patients to reduce the incidence of febrile and urticarial reactions as well as infectious cytomegalovirus contamination. If they are not available, frozen thawed RBCs should be administered. Once a pre transfusion Hb level 9-10 g/dL is achieved, transfusions are administered monthly in infancy and subsequently at 2- to 4-week intervals [49,50]. In clinically stable patients, 8-15 mL RBCs per kilogram of body weight can be infused over a span of 1-2 hours at each transfusion event. If Hb levels are 5 g/dLand/or in the presence of heart failure, smaller aliquots of RBCs (5 mL/kg) should be administered to prevent volume overload until the Hb level is gradually increased to 9 g/dL. A clinical record of all transfusion events should be monitored annually to identify hypersplenism. A record of weight, the amount of blood transfused at each visit, and the pre transfusion Hb level is needed to calculate the annual transfusion requirement [51].
Iron chelation therapy :-
Children with thalassemia major should begin therapy at the earliest possible age and certainly by the time they have accumulated more than 7 g of excess iron. In young children, a serum ferritin level much greater than 1.000 μg/l or 1 year of regular transfusions (or both) can be used as surrogate indicators to initiate chelation therapy [52].
|
Parameter |
Deferoxamine (DFO) |
Deferiprone (DFP) |
Deferasirox (DFX) |
|
Route of administration |
Parenteral (subcutaneous/IV infusion) |
Oral (3 times daily) |
Oral (once daily) |
|
Compliance |
Poor (due to long infusions) |
Moderate |
Good (convenient dosing) |
|
Effect on iron overload |
Effective (especially hepatic iron removal) |
Very effective for cardiac iron |
Effective for both hepatic and cardiac iron |
|
Clinical outcomes |
Improves survival but limited by compliance |
Reduces cardiac complications significantly |
Improves overall iron balance and reduces transfusion-related complications |
|
Quality of life |
Lower (painful infusions, time-consuming) |
Better than DFO (oral route) |
Highest (once daily, easy use) |
|
Major adverse effects |
Growth retardation, ocular/ear toxicity, infections |
Agranulocytosis, GI upset, joint pain |
GI upset, rash, mild renal effects |
|
Monitoring requirement |
High (toxicity monitoring) |
Very high (blood counts) |
Moderate (renal/liver function) |
|
Overall suitability |
Effective but less preferred today |
- |
- |
Table 1. Comparative Evaluation of Iron Chelation Therapies (DFO, DFP, and DFX) in Iron Overload Management
Deferoxamine :-
Deferoxamine (DFO) is a hexadentate iron chelator (deferoxamine mesylate; desferal®). DFO was introduced as parenteral therapy for iron overload associated with β-thalassemia major in 1976 [54]. Plasma half-life of DFO is short (20–30 min). Therefore, standard treatment involves the subcutaneous infusion of 40 mg DFO for 8–12 h nightly for 5–7 nights weekly using a battery-operated infusion pump. Subcutaneous administration is preferred except in patients with severe cardiac iron deposition, for whom continuous intravenous deferoxamine therapy is recommended. Iron excretion occurs through biliary and urinary routes [53].
Adverse events of DFO include growth retardation, skeletal changes, ocular and auditory disturbances, pulmonary, and renal toxicities. They are preventable if proper monitoring is practiced to detect early signs of toxicity. Susceptibility to infection with Yersinia and perhaps other Gram-negative bacilli is increased in thalassemia patients who receive DFO therapy. Painful local skin reactions at the infusion site are common. Zinc deficiency can occur [53].
Deferiprone:-
(DFP) is an orally administered bidentate iron chelator (Ferriprox®, Kelfer®). The usual dose of DFP is 75–100 mg/kg/day taken orally in three divided doses. Plasma half-life of DFP is 2–3 h, and iron is mainly excreted in urine [54]. Adverse effects of DFP include gastrointestinal disturbances, agranulocytosis and neutropenia, arthropathy, increased liver enzyme levels, and low plasma zinc level [53,54].
Deferasirox:-
Deferasirox (DFX) is an orally administered tridentate iron chelator that is indicated for the treatment of transfusion iron overload in persons more than 2 years of age. The US Food and Drug Administration approves a recommended daily dose of 20–40 mg/kg body weight, taken once on an empty stomach at least 30 min before food [53]. The most common adverse events with DFX therapy include gastrointestinal disturbances, rash, and mild increases in serum creatinine [55].
Gene Therapy :-
Murine-thalassemia models have been successfully cured with the use of a retroviral vector (TN39) transferring the human-globin gene sequence and its promoter region into murine stem cells of TI and TMmice [56,57]. Globin gene transfer into progenitor hematopoietic cells of humans is also being studied [58,59]. However, concerns regarding gene transfer include the need for improved efficiency of gene delivery and mastery of vector stability, viral titers, nononcogenic insertion, the variable expression of globin genes, and the variable contributions of the-thalassemia phenotype and other modifiers to the effectiveness of gene transfer [60]. One regularly transfused patient with Hb-E/ o thalassemia has been reported who, after nonmyeloablative conditioning, received autologous bone marrow CD34 cells transduced with a lentiviral vector expressing a A-787Q globin gene, and has remained stable without transfusion support for 2 years [61]. In addition, a phase 1 study of transfusion-dependent-thalassemia patients using the TNS q.3.55 lentiviral vector encoding human-globin gene after nonmyeloablative conditioning is planned. This approach may prevent graft rejection in patients who do not have identically matched HLA donors and therefore are at higher risk to develop GVHD and continuous immune suppression [62]
|
Approach |
Description |
|
Retroviral Vector |
TN39 transferring human-globin gene sequence into murine stem cells |
|
Lentiviral Vector |
TNS q.3.55 encoding human-globin gene |
|
Target Cells |
Murine stem cells, human progenitor hematopoietic cells |
|
Delivery Method |
Gene transfer into CD34 cells |
|
Conditioning |
Nonmyeloablative conditioning |
|
Outcome |
One patient remained stable without transfusion support for 2 years |
Table 2. Gene Therapy Approaches and Outcomes in β-Globin Gene Transfer Studies
Several other molecular approaches for gene therapy using different mutations of stop codons and aberrant splicing have also been described [60]. Gene therapy is a promising approach to curing thalassemia but is still in the early investigational phase trials. In conclusion, we have tried to describe the different clinical manifestations of thalassemia with the optimal care that is available today. However, very different treatment approaches exist world wide depending on factors, such as socioeconomic conditions, cultural traditions, and the quality of available health care. Currently, in parts of the world where sufficient resources exist to support optimal transfusion and chelation programs, thalassemia patients are living longer and maintaining a good quality of life, with a select few being cured using bone marrow transplantation [51,63].
Folic acid supplementation:-
Folic acid (vitamin B9) supplementation is often prescribed to individuals with thalassemia to support red blood cell production and minimize the risk of developing megaloblastic anemia [64].
|
Parameter |
Folic Acid in Thalassemia |
Future Prospects |
|
Role |
Supports RBC production (erythropoiesis |
May be optimized as part of combination therapy |
|
Reason for use |
Increased folate demand due to rapid RBC turnover |
Better understanding of individual folate needs (personalized dosing) |
|
Indication |
Commonly used in non-transfusion dependent thalassemia |
Targeted use based on disease severity and genotype |
|
Dose |
1–5 mg/day orally |
Dose optimization using biomarkers and patient-specific response |
|
Mechanism |
Aids DNA synthesis and maturation of RBC precursors |
Integration with advanced therapies to enhance erythropoiesis |
|
Clinical benefits |
Helps reduce anemia severity and supports bone marrow activity |
Potential to improve outcomes when combined with new drugs (e.g., gene therapy, erythroid modulators) |
|
Safety |
Safe, well tolerated |
Continued use due to excellent safety profile |
|
Limitations |
Does not treat underlying genetic defect |
Will remain supportive, not curative |
|
Research direction |
Basic supportive therapy |
Focus on personalized medicine and adjunct use with novel treatments |
Table 3. Current Role and Future Prospects of Folic Acid Supplementation in Thalassemia Management
Splenectomy
If the annual red cell requirement exceeds 180-200 ml/Kg of RBC (assuming that the Hct of the unit of red cells is about 75%), splenectomy should be considered, provided that other reasons for increased consumption, such as hemolytic reactions, have been excluded. Other indications for splenectomy are symptoms of splenic enlargement, leukopenia and/or thrombocytopenia and increasing iron overload despite good chelation [65].
CONCLUSION
Thalassemia remains a complex and multifaceted genetic disorder with far-reaching implications for individuals, families, and healthcare systems globally. Despite advancements in understanding and treating the condition, significant challenges persist, underscoring the need for sustained efforts to combat this disease. A collaborative effort from healthcare providers, policymakers, researchers, and advocacy groups is essential to address these challenges effectively. This collective approach can help bridge gaps in care, facilitate access to innovative treatments, and support those affected by thalassemia.Managing thalassemia requires a continuum of care from early detection and screening to comprehensive treatment and innovative therapies like gene therapy. Implementing universal screening programs, ensuring access to quality care, and promoting awareness are crucial steps in this journey. While progress has been made in areas like iron chelation therapy and gene therapy, there is still much work to be done to ensure equitable healthcare access, reduce disease burden, and support the psychosocial well-being of individuals living with thalassemia and their families.The path forward involves continued research, improved healthcare strategies, and robust support systems. By working together, we can strive for better patient outcomes, enhanced quality of life, and ultimately, a cure for this chronic condition. Addressing thalassemia effectively will not only benefit those directly affected but also contribute to broader advancements in genetic disorder management and healthcare innovation.
REFERENCES





Krutika Agale1*, Sushant Fadtare2, Srushti Kamble1, Gayatri Chile1, Namrata Jadhav1, Precision Management Of Iron Overload: Navigating Evolving Diagnostic And Therapeutic Paradigms In Β-Thalassemia., Int. J. Sci. R. Tech., 2026, 3 (5), 966-979. https://doi.org/10.5281/zenodo.20411267
 10.5281/zenodo.20411267
10.5281/zenodo.20411267