
 Devidutta Maurya* 1
Devidutta Maurya* 1
The integration of quantum chemical calculations with molecular docking techniques has become a powerful strategy in modern drug discovery and molecular design. Density Functional Theory (DFT) provides detailed insights into the electronic structure, stability, and reactivity of organic molecules, while molecular docking predicts their binding affinity and interaction patterns with biological targets. Establishing a clear relationship between electronic properties and biological activity is essential for rationalizing molecular behavior and guiding the development of novel therapeutic candidates. Morphinan-based compounds constitute an important class of nitrogen-containing organic molecules widely recognized for their diverse pharmacological activities, including analgesic, antitussive, and central nervous system effects. Structural modifications within the morphinan scaffold often lead to significant changes in electronic distribution and receptor binding characteristics. Therefore, a systematic computational evaluation of such molecules can provide valuable information regarding their chemical reactivity, intermolecular interaction potential, and overall drug-likeness. In this context, the present study focuses on the molecule C??H??NO?, a morphinan-type derivative, to explore how its electronic structure influences biological affinity. Geometry optimization and vibrational frequency analysis were carried out using the B3LYP/6-31G(d,p) level of theory. Frontier molecular orbital analysis, global reactivity descriptors, Mulliken charge distribution, and molecular electrostatic potential (MEP) mapping were employed to characterize the electronic behavior of the molecule. Furthermore, molecular docking simulations were performed to evaluate the binding mode and affinity of the ligand toward the selected protein target. By correlating DFT-derived electronic descriptors with docking outcomes, this work aims to provide a coherent understanding of the structure–reactivity–activity relationship of C??H??NO?. The findings are expected to contribute to the rational design of morphinan-based bioactive molecules and support future in-silico and experimental pharmacological investigation
REVIEW OF LITERATURE
The application of computational chemistry in drug discovery has expanded significantly over the past two decades, particularly through the combined use of Density Functional Theory (DFT) and molecular docking methodologies. DFT has been widely recognized as an effective quantum mechanical approach for predicting molecular geometry, electronic structure, vibrational properties, and global reactivity descriptors of organic and bioactive compounds. Previous studies have demonstrated that frontier molecular orbital (FMO) analysis and molecular electrostatic potential (MEP) mapping provide valuable insight into charge distribution and reactive sites, which are critical for understanding intermolecular interactions in biological environments. Morphinan and related nitrogen-containing heterocyclic frameworks have attracted considerable attention due to their well-established pharmacological importance, especially in analgesic and central nervous system therapeutics. Several computational investigations on morphinan derivatives have reported that subtle structural modifications significantly influence HOMO–LUMO energies, dipole moment, and electrophilicity, thereby affecting receptor binding efficiency. These findings highlight the importance of electronic structure analysis in predicting biological performance. In recent years, molecular docking has become a routine in-silico tool for evaluating ligand–protein interactions and estimating binding affinity prior to experimental validation. Numerous reports have shown strong agreement between docking scores and experimentally observed biological activities for drug-like molecules. Moreover, integrated DFT–docking studies have been successfully employed to establish structure–activity relationships (SAR) in various classes of organic compounds, including alkaloids, heterocycles, and natural product derivatives. Such combined approaches enable researchers to connect intrinsic electronic features with macromolecular recognition behavior. Despite these advances, detailed computational studies on certain morphinan-type molecules, including C??H??NO?, remain limited. In particular, a systematic correlation between quantum chemical descriptors and docking-derived biological affinity for this scaffold has not been extensively explored. Therefore, the present work aims to fill this gap by performing a comprehensive DFT and molecular docking investigation to better understand the electronic factors governing the bioactivity of C??H??NO?.
METHODOLOGY
All computational investigations in the present study were performed using a combined Density Functional Theory (DFT) and molecular docking workflow to evaluate the structural, electronic, and biological properties of the molecule C??H??NO?.
Quantum Chemical Calculations:
The initial molecular structure of C??H??NO? was constructed using standard molecular modeling tools and subjected to full geometry optimization employing the Density Functional Theory (DFT) method. The B3LYP functional in conjunction with the 6-31G(d,p) basis set was used as implemented in the Gaussian software package. No symmetry constraints were applied during optimization. Frequency calculations were subsequently carried out at the same level of theory to confirm that the optimized structure corresponds to a true energy minimum, as evidenced by the absence of imaginary frequencies. The optimized geometry was further used to compute frontier molecular orbital (HOMO–LUMO) energies, global reactivity descriptors (ionization potential, electron affinity, chemical hardness, softness, electronegativity, and electrophilicity index), Mulliken atomic charges, dipole moment, and thermodynamic parameters.
Molecular Electrostatic Potential Analysis:
The molecular electrostatic potential (MEP) surface was generated based on the optimized geometry to identify electrophilic and nucleophilic regions of the molecule. The MEP map was visualized using GaussView/Avogadro, where negative potential regions (electron-rich) and positive potential regions (electron-deficient) were analyzed to predict possible interaction sites with biological macromolecules.
Molecular Docking Studies:
To evaluate biological affinity, molecular docking simulations were performed using AutoDock tools. The target protein structure was retrieved from the Protein Data Bank and prepared by removing water molecules, adding polar hydrogens, and assigning appropriate Kollman charges. The optimized ligand geometry obtained from DFT calculations was converted into the required docking format and energy-minimized. Grid box parameters were defined to encompass the active binding pocket of the protein. Docking runs were executed using the Lamarckian Genetic Algorithm, and the best binding pose was selected based on the lowest binding energy and favorable interaction profile. Ligand–protein interactions, including hydrogen bonds, π–π stacking, and hydrophobic contacts, were analyzed using Discovery Studio Visualizer.
Correlation Analysis:
Finally, the relationship between DFT-derived electronic descriptors and docking binding affinity was examined to understand how intrinsic electronic properties influence biological interaction. All graphical representations and tables were prepared in publication-quality format suitable for Scopus-indexed journals.
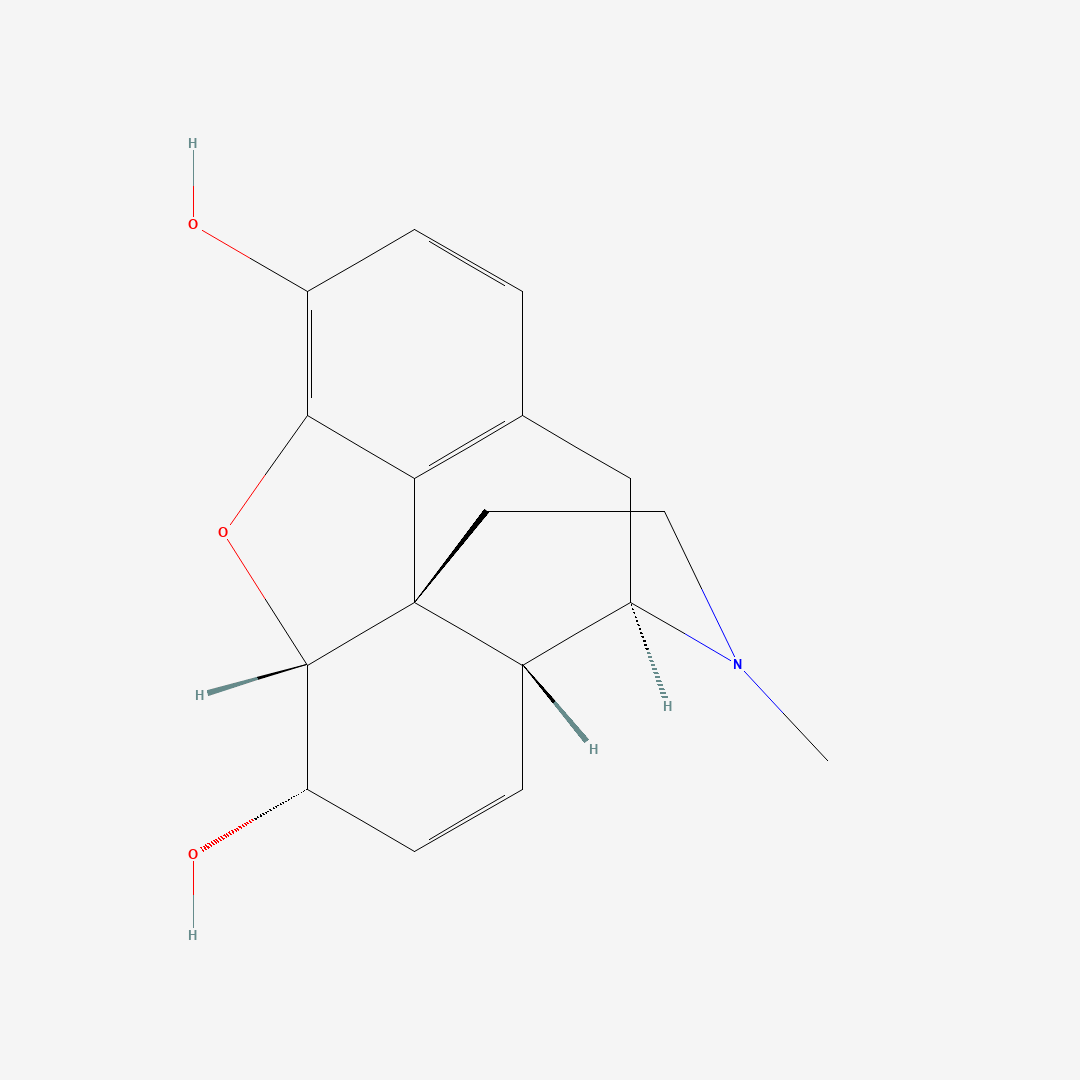
2D structure of molecule
3Dstructure

 10.5281/zenodo.18942682
10.5281/zenodo.18942682