
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Shri Shankaracharya Institute of Pharmaceutical Sciences and Research, Bhilai, Chhattisgarh, India, 490020
Breast cancer remains a major global health challenge and a leading cause of cancer-related mortality among women, with hormone-dependent progression primarily mediated by estrogen biosynthesis. Aromatase, a key enzyme in estrogen production, represents a validated therapeutic target; however, the clinical utility of prevailing inhibitors is often limited by resistance and long-term toxicity. Thus, the development of novel, potent, and selective aromatase inhibitors is of significant interest. In the present study, a series of structurally diverse 1,2,4-triazole derivatives (4a-f) was rationally designed and efficiently synthesized via a microwave-assisted protocol. The synthesized compounds were comprehensively characterized by spectroscopy methods. Their antioxidant potential was evaluated using the DPPH assay, while anticancer activity was evaluated against the MCF-7 human breast cancer cell line using the MTT assay. Moreover, molecular docking studies were conducted employing Molegro Virtual Docker against human placental aromatase (PDB ID: 3S79) to illuminate binding affinity and interaction patterns. Biological evaluation revealed that compound 4c exhibited the most potent antioxidant activity (IC50 = 24.78 ± 0.87 µM) and pronounced cytotoxic effects, followed by compound 4b. SAR analysis indicated that substituents that donate electrons greatly improve biological performance. Docking analysis confirmed favorable binding profiles for all derivatives, with compound 4c displaying the lowest MolDock score (-151.841 kcal/mol), outperforming the letrozole. These results show that compound 4c is a promising lead candidate with better antioxidant and anticancer properties, as shown by its strong in silico binding affinity. This study underscores the therapeutic promise of triazole scaffolds in the development of next-generation anti-breast cancer agents.
Breast cancer is one of the most commonly diagnosed malignancies and a leading cause of cancer-related mortality among women worldwide, representing a major global health burden. According to GLOBOCAN 2020, approximately 2.3 million new cases were reported, highlighting its high incidence and clinical significance [1,2]. Although there has been persistent progress in measures of diagnosis and treatment, the long-term management of breast cancer has been reported to be a challenge because of the emergence of drug-resistant strains, adverse effects, and recurrence of the tumor, in effect, leading to poor clinical outcomes. These shortcomings underline the necessity of the creation of new therapeutic agents that are more effective and less resistant. The reactions of a significant number of breast cancers are hormone-dependent and dependent on estrogen to develop and progress [3]. Aromatase is a cytochrome P450 enzyme that is pivotal in the production of estrogen, making it a key therapeutic target. Aromatase inhibition has already become a successful approach to the management of hormone-sensitive breast cancer, especially in postmenopausal women [4]. Aromatase inhibitors that have been used clinically, like Anastrozole, Letrozole, and Exemestane, have shown therapeutic effects. Nevertheless, their chronic application is usually constrained by their diminished selectivity, adverse side effects, and development of resistance, which has compelled exploration of fresh scaffolds with improved pharmacological characteristics [5].
Nitrogen-containing heterocyclic molecules, especially 1,2,4-triazole derivatives, are of special interest to medicinal chemistry because of their wide range of biological actions and good pharmacokinetic characteristics. The 1, 2, 4 - triazole nucleus is believed to be a privileged scaffold because of the high stability rate of the nucleus with respect to metabolism, great aptitude to bind a hydrogen bond, and its capacity to coordinate the heme iron of a particular aromatase enzyme, hence inhibiting the activity. There has been widespread reporting of triazole-based compounds possessing antimicrobial, anti-inflammatory, and anticancer properties [6,7]. Interestingly, other triazole analogs have exhibited good aromatase-inhibitory properties, which justifies their use in the treatment of breast cancer. This analysis focuses on the logical design and synthesis of a new group of 1,2,4-triazole compounds and their evaluation for anticancer properties. The triazole scaffold was selectively subjected to structural changes to increase binding affinity against the aromatase enzyme and to enhance the pharmacokinetic changes. In addition, structure-activity relationship (SAR) was used to explain the effect of various substituents on biological activity, as well as the use of lead compounds. This study provides valuable insights into the development of new triazole-based aromatase inhibitors as promising candidates for the treatment of hormone-dependent breast cancer.
2. MATERIALS AND METHODS
2.1 Chemicals
All chemicals and reagents were of analytical grade and procured from Sigma-Aldrich, Merck, SRL Chemicals, Loba Chemie, and CDH Chemicals. Reaction progress was monitored by TLC on silica gel G plates. 1H and 13C NMR spectra were recorded on a Bruker Advance Neo 400 MHz spectrometer using DMSO-d6. FTIR spectra were obtained using a PerkinElmer Spectrum IR. Mass spectra were recorded on an Xevo G2-XS QT quadrupole time-of-flight mass spectrometer, and elemental analysis was performed using a Thermo Scientific Flash 2000 analyzer.
2.2 Chemistry
2.2.1 General procedure for the synthesis of 1H-1,2,4-triazol-3-amine (1)
Aminoguanidine (0.01 mol) and formic acid (0.01 mol) were dissolved in ethanol (10 mL) in the presence of KOH and subjected to microwave irradiation (280 W, 60-65 °C) in two runs for 10 minutes. After completion, the reaction mixture was cooled, neutralized with dilute HCl, and acetone was added to induce precipitation. The resulting solid was filtered, washed with cold acetone, and dried to obtain the desired compound.
IR (KBr, cm-1): 3352.44 (N-H), 3297.74 (N-H), 1645.68 (C=N), 1550.44 (N-N), 1267.35 (C-N); 1H NMR (400 MHz, DMSO-d6) ẟppm: 5.57-5.87, 7.79-8.07, 10.87-11.32; 13C NMR (400 MHz, DMSO-d6) ẟppm: 147.80, 152.44.
2.2.2 General procedure for the synthesis of 4-(((1H-1,2,4-triazol-3-yl) imino) methyl) benzonitrile (2)
Compound 1 (0.01 mol) and cyanobenzaldehyde (0.01 mol) were dissolved in ethanol with a catalytic amount of acetic acid and subjected to microwave irradiation (280 W, 50-60 °C) for 15 minutes. After completion, crushed ice was added, and the mixture was left to stand for 30 minutes to allow precipitation. The solid product was then filtered, washed with cold water or ethanol, and dried to obtain the desired compound.
IR (KBr, cm-1): 3045.74 (Ar C-H), 2232.69 (C≡N), 1644.82 (C=N), 1586.41 (C=C), 1548.59 (N-N), 1234.62 (C-N); 1H NMR (400 MHz, DMSO-d6) ẟppm: 7.21-7.32, 7.43-7.61, 7.71-7.92, 8.20-8.42, 8.53-8.74, 11.07-11.40; 13C NMR (400 MHz, DMSO-d6) ẟppm: 112.82, 118.35, 121.41, 126.16, 128.62, 130.50, 136.71, 145.84, 149.61, 152.33.
2.2.3 General procedure for the synthesis of 4-(((1-(3-oxobutyl)-1H-1,2,4-triazol-3-yl) imino) methyl) benzonitrile (3)
The compound (0.01 mol) was dissolved in acetone with formaldehyde, and 10% KOH was added as a base catalyst. The mixture was subjected to microwave irradiation (280 W, 50-60 °C) for 20 minutes. After cooling, it was neutralized with dilute HCl and poured onto crushed ice to precipitate the product. The solid was filtered, washed with cold water, and dried to obtain the desired compound.
IR (KBr, cm-1): 3064.72 (Ar C-H), 2862.19 (Alp C-H), 2228.34 (C≡N), 1664.80 (C=O), 1671.45 (C=C), 1560.61 (C=N), 1259.26 (C-N); 1H NMR (400 MHz, DMSO-d6) ẟppm: 2.04-2.15, 2.60-2.79, 3.18-3.40, 7.52-7.71, 7.77-7.95, 8.18-8.40, 8.51-8.71; 13C NMR (400 MHz, DMSO-d6) ẟppm: 29.85, 34.70, 41.55, 112.92, 118.46, 121.33, 126.42, 128.91, 131.03, 145.70, 149.35, 152.24, 197.60.
2.2.4 General procedure for the synthesis of N'-(4-(3-((substitutedbenzylidene) amino)-1H-1,2,4-triazol-1-yl) butan-2-ylidene) isonicotinohydrazide (4a-f)
Compound 3 (0.01 mol) and the appropriate hydrazide derivative (0.01 mol) were dissolved in ethanol as a solvent in the presence of a catalytic amount of glacial acetic acid. The reaction mixture was transferred to a microwave-compatible vessel and irradiated at 350 W (60-70 °C) for 15-20 minutes. After completion, the mixture was cooled to room temperature, neutralized with dilute HCl, and poured onto crushed ice to induce precipitation. The resulting solid was filtered, washed with cold water or ethanol, and dried to obtain the desired compound.
2.2.4.1 General procedure for the synthesis of N'-(4-(3-((4-cyanobenzylidene) amino)-1H-1,2,4-triazol-1-yl) butan-2-ylidene) isonicotinohydrazide (4a)
Color: yellow powder; % Yield: 82.45; M.P.: 81-83 °C; IR (KBr, cm-1): 3365.41 (N-H), 3062.74 (Ar C-H), 2925 (Alp C-H), 2226.45 (C≡N), 1665.74 (C=O), 1618.40 (C=N), 1565.74 (Ar C=C); 1H NMR (400 MHz, DMSO-d6) ẟppm: 2.11, 2.61, 4.02, 7.54, 7.81, 7.98, 8.77. 9.53, 10.86; 13C NMR (400 MHz, DMSO-d6) ẟppm: 19.8, 29.5, 42.1,114.8, 118.4, 121.7, 126.1, 132.2, 140.6, 143.7, 149.4, 158.1, 163.1; MS: calculated [M+] m/z Found: 386.12; Elemental Analysis Found: C, 62.11; H, 4.68; N, 28.98; O, 4.11.
2.2.4.2 General procedure for the synthesis of 4-(((1-(3-(2-phenylhydrazineylidene) butyl)-1H-1,2,4-triazol-3-yl)imino)methyl) benzonitrile (4b)
Color: off white powder; % Yield: 83.54; M.P.: 80-84 °C; IR (KBr, cm-1): 3365.85 (N-H), 3072.85 (Ar C-H), 2942.74 (Alp. C-H), 2234.52 (C≡N), 1618.72 (C=N), 1561.25 (Ar C=C); 1H NMR (400 MHz, DMSO-d6) ẟppm: 1.93, 2.60, 4.01, 7.05, 7.32, 7.53, 7.98, 8.76 8.97; 13C NMR (400 MHz, DMSO-d6) ẟppm: 14.1, 23.7, 42.0, 113.9, 114.6, 118.6, 122.3, 126.2, 132.2, 140.5, 143.7, 158.1, 158.4; MS: calculated [M+] m/z Found: 357.05; Elemental Analysis Found: C, 67.15; H, 5.22; N, 27.34.
2.2.4.3 General procedure for the synthesis of N'-(4-(3-((4-cyanobenzylidene) amino)-1H-1,2,4-triazol-1-yl) butan-2-ylidene)-4-methylbenzohydrazide (4c)
Color: white powder; % Yield: 79.85; M.P.: 85-88 °C; IR (KBr, cm-1): 3365.81 (N-H), 3069.75 (Ar C-H), 2922.75 (Alp. C-H), 2221.45 (C≡N), 1661.70 (C=O), 1618.45 (C=N), 1526.74 (Ar C=C); 1H NMR (400 MHz, DMSO-d6) ẟppm: 1.92, 2.40, 2.60. 4.01, 7.30, 7.52, 7.82, 7.98, 9.52, 10.82; 13C NMR (400 MHz, DMSO-d6) ẟppm: 10.0, 13.7, 31.4, 42.4, 114.4, 118.1, 126.2, 140.6, 143.6, 158.2, 160.4; MS: calculated [M+] m/z Found: 399.11; Elemental Analysis Found: C, 66.10; H, 5.22; N, 24.51; O, 4.00.
2.2.4.4 General procedure for the synthesis of N'-(4-(3-((4-cyanobenzylidene) amino)-1H-1,2,4-triazol-1-yl) butan-2-ylidene) propionohydrazide (4d)
Color: white powder; % Yield: 76.15; M.P.: 80-83 °C; IR (KBr, cm-1): 3361.78 (N-H), 3061.85 (Ar C-H), 2929.54 (Alp. C-H), 2232.15 (C≡N), 1653.16 (C=O), 1617.19 (C=N), 1532.74 (Ar C=C); 1H NMR (400 MHz, DMSO-d6) ẟppm: 1.01, 1.90, 2.26, 2.60, 7.53, 7.94, 8.73, 9.54, 10.53; 13C NMR (400 MHz, DMSO-d6) ẟppm: 10.2, 13.4, 31.5, 42.0, 114.5, 118.2, 126.2, 140.5, 143.7, 158.0, 160.2; MS: calculated [M+] m/z Found: 337.17; Elemental Analysis Found: C, 60.44; H, 5.58; N, 29.01; O, 4.62.
2.2.4.5 General procedure for the synthesis of 4-amino-N'-(4-(3-((4-cyanobenzylidene) amino)-1H-1,2,4-triazol-1-yl) butan-2-ylidene) benzohydrazide (4e)
Color: white powder; % Yield: 76.82; M.P.: 82-86 °C; IR (KBr, cm-1): 3362.41 (N-H), 3452.45 (-NH2), 3062.75 (Ar C-H), 2921.50 (Alp. C-H), 2224.40 (C≡N), 1661.07 (C=O), 1634.41 (C=N), 1567.15 (Ar C=C); 1H NMR (400 MHz, DMSO-d6) ẟppm: 1.90, 2.60, 4.01, 5.44, 6.50, 7.52, 7.53, 9.54, 10.44; 13C NMR (400 MHz, DMSO-d6) ẟppm: 13.7, 42.0, 114.2, 118.6, 122.3, 126.7, 132.1, 151.3, 158.2, 160.4; MS: calculated [M+] m/z Found: 400.11; Elemental Analysis Found: C, 62.85; H, 5.01; N, 27.72; O, 3.85.
2.2.4.6 General procedure for the synthesis of N'-(4-(3-((4-cyanobenzylidene) amino)-1H-1,2,4-triazol-1-yl) butan-2-ylidene) formohydrazide (4f)
Color: white powder; % Yield: 80.75; M.P.: 84-86 °C; IR (KBr, cm-1): 3349.75 (N-H), 3063.04 (Ar C-H), 2925.74 (Alp. C-H), 2224.40 (C≡N), 1682.85 (C=O), 1620.74 (C=N), 1581.72 (Ar C=C); 1H NMR (400 MHz, DMSO-d6) ẟppm: 1.91, 2.60, 7.52, 7.97, 8.19, 8.76, 9.52, 10.84; 13C NMR (400 MHz, DMSO-d6) ẟppm: 13.5, 23.5, 42.01, 117.5, 125.6, 132.1, 143.6, 158.1, 166.0; MS: calculated [M+] m/z Found: 309.12; Elemental Analysis Found: C, 58.20; H, 4.73; N, 31.65; O, 5.12
3. BIOLOGICAL ACTIVITY
3.1 Antioxidant activity
The DPPH (2, 2-diphenyl- 1-picrylhydrazyl) free radical scavenging test assessed the ability of the synthesized compounds (4a-f) to act as antioxidants. Each compound was made up (10 mM into stock solutions in ethanol and then diluted to form a concentration range of 5-25 µM/mL. In the case of assay, 0.1 mL of each test solution was mixed with 3 mL of a 0.1 mM solution of DPPH. The mixtures of the reaction were kept in the dark at room temperature to allow a full interaction between the antioxidant compounds and DPPH radicals. A UV-Visible spectrophotometer was used to measure the decrease in absorbance at 517 nm. A control was made using the DPPH solution in place of the test sample using DMSO. The radical scavenging activity was determined by associating the absorbance of the test samples with that of the control [8].
3.2 Cytotoxicity study
The MTT assay of the cytotoxic activity of the compounds (4a-f) was done on MCF-7 human cancer cell lines. Cells were seeded at a cell density of 2.5 thinly sliced cells/well (or 2.5 lives/plate) in 96-well plates, incubated at 370 C in a moist 5% CO2 environment to allow the cells to bond. Washing in PBS was done, followed by treatment of cells with various concentrations of the compounds (200 nM, 2, 5, 10, and 20 nM) and 72h of incubation. Then 10 µL of MTT solution was put in each well and incubated for 4 h in order to allow the formation of formazan crystals. They dissolved the crystals with 10% SDS in 0.01 N HCl, and the absorbance was measured at 562 nm by a microplate reader, and cell viability was determined against an untreated control [9].
4. Computational study
4.1 Molecular Docking Simulation
Molecular docking analyses were conducted to understand the binding affinities and binding profiles of the prepared triazole analogs (4a-f) with human placenta aromatase cytochrome P450. Two-dimensional (2D) chemical representations of all target compounds, as well as a reference inhibitor, Letrozole, were generated in ChemDraw 22.2.0. Energy minimization of these structures was then done with the help of the MM2 force field to get the most thermodynamically stable conformational structures. The optimized geometries were then transformed to three-dimensional (3D) format and stored as 3D PDB files to be used in subsequent computational analysis [10]. Human placental aromatase is a crystallographic structure that was obtained at The RCSB Protein Data Bank (PDB ID: 3S79; resolution: 2.75 A) [11]. Protein preparation consisted of all crystallographic water molecules and co-crystallized ligands removed, and necessary hydrogen atoms added to stabilize the structure to use in docking simulations. Molegro Virtual Docker v6.0 was used to carry out molecular docking simulations, which included an advanced cavity detection algorithm that was used to identify potential ligand-binding sites. The grid box dimension was set at 30 X30 X30 A, centred around the active site region, being X: -0.92, Y: 52.67, and Z: 6.75 with a radius of 15.0 A about the active site region. Docking resolution was set to 0.30 A, to provide accuracy in the positioning of the ligand. To promote the reliability and strength of the docking results, every ligand was docked 10 times in separate docking replicates, using a population size of 50 and a maximum of 1500 docking iterations per replicate, thus allowing exhaustive conformational sampling. The highest docking poses were chosen using binding energy values and stability of the interactions [12]. Lastly, the binding orientations, intermolecular interactions, and amino acid residues of the active site that bind to the ligand were carefully analyzed and visualized with BIOVIA Discovery Studio Visualizer [13].
3. RESULTS AND DISCUSSION
3.1 Chemistry
The synthesized compounds (4a-f) were successfully confirmed by spectral and analytical data, demonstrating the formation of the targeted triazole-based derivatives. Compound (1) exhibited characteristic IR peaks at 3352.44 and 3297.74 cm⻹ (N-H), 1645.68 cm⻹ (C=N), 1550.44 cm⻹ (N-N), and 1267.35 cm⻹ (C-N), while 1H NMR signals at δ 5.57-5.87, 7.79-8.07, and 10.87-11.32 ppm supported the presence of NH and heteroaromatic protons, further confirmed by ¹³C NMR signals at δ 147.80 and 152.44 ppm. Compound (2) showed absorption bands at 3045.74 cm⻹ (Ar C-H), 2232.69 cm⻹ (C≡N), 1644.82 cm⻹ (C=N), and 1586.41 cm⻹ (C=C), with ¹H NMR signals in the aromatic region δ 7.21–8.74 ppm and NH at δ 11.07-11.40 ppm, while ¹³C NMR signals (δ 112.82-152.33 ppm) confirmed aromatic and nitrile carbons. Compound (3) displayed peaks at 3064.72 cm⻹ (Ar C–H), 2862.19 cm⻹ (aliphatic C-H), 2228.34 cm⻹ (C≡N), 1664.80 cm⻹ (C=O), and 1560.61 cm⻹ (C=N), with ¹H NMR signals at δ 2.04-3.40 ppm (aliphatic) and δ 7.52-8.71 ppm (aromatic), and a characteristic carbonyl carbon at δ 197.60 ppm in ¹³C NMR. The final derivatives (4a-f) consistently exhibited IR bands in the range 3349.75-3365.85 cm⻹ (N-H), 3061.85-3072.85 cm⻹ (Ar C-H), 2921.50-2942.74 cm⻹ (aliphatic C-H), 2221.45-2234.52 cm⻹ (C≡N), 1653.16-1682.85 cm⻹ (C=O), 1617.19-1634.41 cm⻹ (C=N), and 1526.74-1581.72 cm⻹ (Ar C=C), confirming the presence of key functional groups. The 1H NMR spectra revealed signals for aliphatic protons (δ 1.01-4.02 ppm), aromatic protons (δ 7.05-8.77 ppm), and -NH protons (δ 9.52-10.86 ppm), while 13C NMR data (δ 10.0-166.0 ppm) further supported the structural framework. Mass spectra showed molecular ion peaks consistent with calculated values (m/z 309.12-400.11), and elemental analysis data were in close agreement with theoretical compositions, confirming the effective synthesis and purity of the compounds.
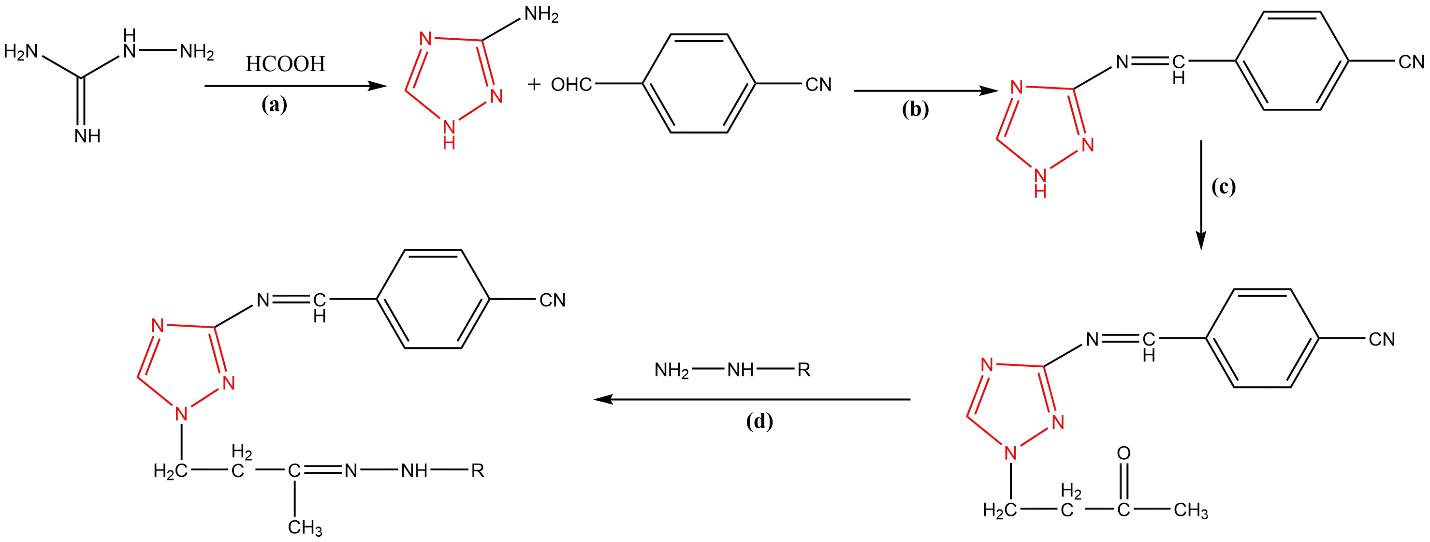
Scheme 1. Synthetic strategies of novel 1,2,4-triazole derivatives (4a-f).
Reaction Conditions: KOH, ethanol, 280 W, 60-65 °C, reflux, 10 minutes, (b) Ethanol, acetic acid, 280 W, 50-60 °C, reflux, 15 minutes, (c) 10% KOH, microwave irradiation 280 W, 50-60 °C, reflux, 20 minutes, (d) glacial acetic acid, 350 W, 60-70 °C, reflux, 15-20 minutes.
|
Compound ID |
R |
|
4a |
|
|
4b |
|
|
4c |
|
|
4d |
|
|
4e |
|
|
4f |
|
3.2 Biological Activity
3.2.1 Antioxidant activity
The antioxidant activity of the synthesized 1,2,4-triazole derivatives (4a-f) was assessed using the DPPH free radical scavenging assay, with Ascorbic acid as the standard (Table 1). The IC50 values indicated that compounds 4c (24.78 ± 0.87 µM) and 4b (28.51 ± 0.52 µM) exhibited superior antioxidant activity compared to the reference drug (32.05 ± 1.29 µM), followed by 4a (30.47 ± 0.61 µM), while compounds 4d (48.44 ± 1.54 µM), 4e (54.03 ± 2.11 µM), and 4f (57.73 ± 2.07 µM) exhibited comparatively lower activity (Figure 1). Structure-activity relationship (SAR) analysis revealed that the enhanced activity of compounds 4c, 4b, and 4a can be attributed to the presence of electron-donating substituents, which increase electron density on the aromatic system and facilitate effective hydrogen or electron donation to stabilize the DPPH radical via resonance. In contrast, derivatives bearing electron-withdrawing groups (4a-f) showed reduced radical scavenging potential due to reduced electron density, limiting their ability to neutralize free radicals. Overall, these findings highlight the crucial role of electronic effects in modulating the antioxidant efficiency of the triazole scaffold.
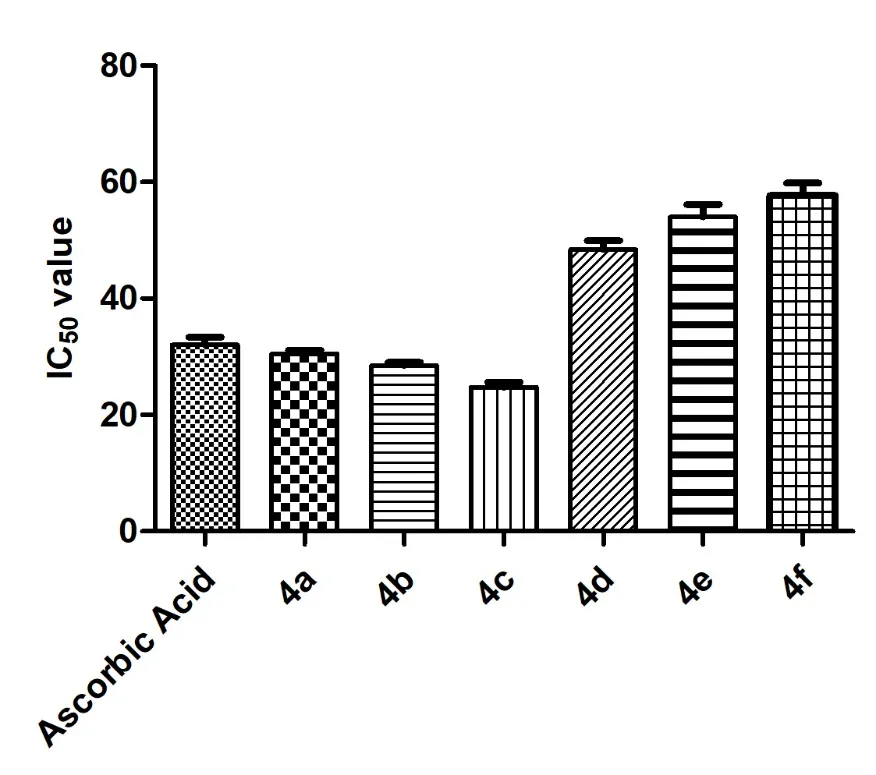
Figure 1. Antioxidant activity of 1,2,4-triazole derivatives (4a-f).
|
Compound ID |
IC50 Valuea |
|
Ascorbic acid |
32.05 ± 1.29 |
|
4a |
30.47 ± 0.61 |
|
4b |
28.51 ± 0.52 |
|
4c |
24.78 ± 0.87 |
|
4d |
48.44 ± 1.54 |
|
4e |
54.03 ± 2.11 |
|
4f |
57.73 ± 2.07 |
Table 1. Antioxidant activity of 1,2,4-triazole derivatives (4a-f).
a IC50 Value expressed in µM±SEM.
3.2.2 Cytotoxicity Study
The cytotoxic activity of the synthesized 1,2,4-triazole derivatives (4a–f) against the MCF-7 breast cancer cell line revealed a clear structure-activity relationship (SAR) influenced by the nature of substituents on the aromatic ring. Among all derivatives, compound 4c exhibited the highest anticancer activity, showing a marked reduction in cell viability even at 200 nM, which may be attributed to the presence of a methyl-substituted aromatic ring (electron-donating group) that enhances electron density, thereby improving interaction with intracellular targets and promoting apoptotic pathways. Compound 4b, bearing a phenyl group, also established significant cytotoxicity with activity comparable to the standard drug Letrozole, suggesting favorable hydrophobic and π-π interactions with biological macromolecules. Compound 4a showed moderate activity, likely due to the presence of a heteroaromatic moiety, which contributes to balanced electronic and steric effects. In contrast, compounds 4d and 4f, containing aliphatic substituents (propionyl and formyl groups), exhibited reduced cytotoxicity, indicating that the absence of aromatic conjugation diminishes binding affinity and cellular uptake (Figure 2). Compound 4e, despite having an -NH2 group (electron-donating), showed comparatively lower activity, possibly due to increased polarity affecting membrane permeability. Overall, the results suggest that electron-donating and aromatic substituents significantly increase anticancer activity, while aliphatic or highly polar groups reduce efficacy. These findings highlight compound 4c as the most promising candidate, with 4b also emerging as a potential lead for further optimization in anticancer drug development.
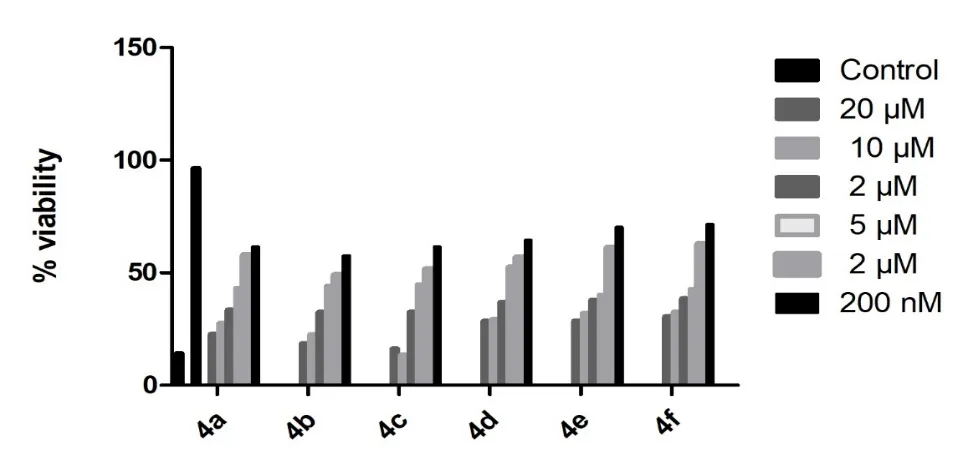
Figure 2. Cytotoxicity study of 1,2,4-triazole derivatives (4a-f).
3.3 Computation Study
3.3.1 Molecular Docking Analysis
Molecular docking studies using Molegro Virtual Docker version 6.0 revealed that the synthesized 1,2,4-triazole derivatives (4a-f) displayed strong binding affinity toward Human placental aromatase (PDB ID: 3S79), a key enzyme involved in estrogen biosynthesis and breast cancer progression. The docking results (Table 2) exhibited MolDock scores ranging from -121.952 to -151.841 kcal/mol, with compound 4c displaying the highest binding affinity (-151.841 kcal/mol), surpassing the reference drug Letrozole (-132.837 kcal/mol). The enhanced binding is attributed to stabilizing interactions such as hydrogen bonding, π-π stacking, and sulfur interactions with crucial amino acid residues. Notably, compound 4c formed multiple hydrogen bonds with Trp141, Ser314, Cys437, and Gly439, along with hydrophobic interactions involving Ile132 and Arg435, conducive to complex stability. Also, compound 4b displayed interaction patterns comparable to Letrozole, particularly with Ser478 and Glu483, as demonstrated in Figure 3. Significantly, residues such as Ser314, Cys437, Trp141, and Glu483 are essential for aromatase activity, and their inhibition may suppress estrogen production, a key strategy in hormone-dependent breast cancer treatment. Molecular visualization of the protein-ligand complex of compound 4b and the standard drug is represented in Figure 4. illustration that the synthesized compounds possess promising potential as aromatase inhibitors, warranting further biological evaluation.
|
Compound ID |
MolDock Score (kcal/mol) |
Interaction (kcal/mol) |
H-Bond (kcal/mol) |
H-bonds Interaction |
S-bonds Interaction |
|
Letrozole |
-132.837 |
-161.029 |
-4.86701 |
Ser478, Glu483 |
Arg192, Gln218, Phe221, Pro308 |
|
4a |
-146.743 |
-119.474 |
-6.26471 |
Trp141, Ser314, Cys437, Ala438, Arg435, |
lle132, Cys437, |
|
4b |
-149.465 |
-85.9401 |
-6.08579 |
Ser314, His402, Cys310, Glu483 |
Gln218, Ser314, |
|
4c |
-151.841 |
-122.876 |
-7.55549 |
Trp141, Ser314, Cys437, Gly439 |
lle132, Trp141, Ser314, Arg435 |
|
4d |
-121.952 |
-110.348 |
-5.37839 |
Ser314, Cys437, Gly439 |
Phe317, Ser363, Val369 |
|
4e |
-129.063 |
-32.2228 |
-4.45422 |
Ser314, His402 Cys437, Gln367 |
Pro368, lle398, Ser363, Val369 |
|
4f |
-136.413 |
-73.5388 |
-7.81719 |
Ser314, His402 |
Thr310, Ser314, Pro368, lle398, Ser363, Val369 |
Table 2. Molecular docking simulations of designed 1,2,4-triazole derivatives.
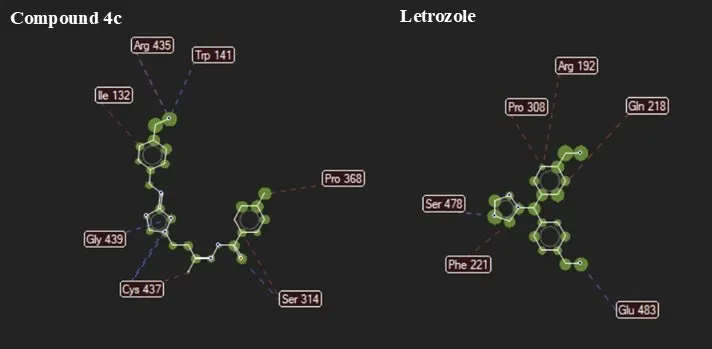
Figure 3. Comparative binding interactions of compound 4c and the reference drug Letrozole within the active site of the target protein.
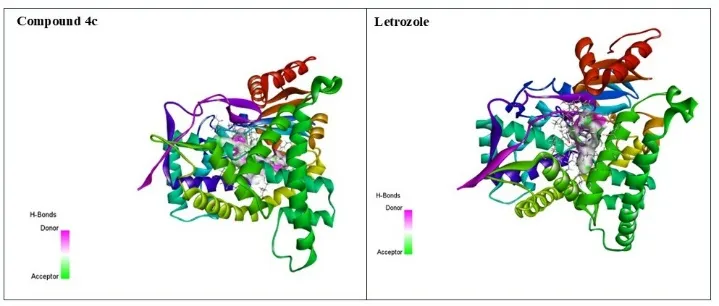
Figure 4. Three-dimensional visualization of the docked poses of compound 4c and the reference drug Letrozole within the active site of the target protein.
CONCLUSION
A novel series of 1,2,4-triazole derivatives (4a-f) was effectively synthesized and structurally confirmed using spectral and analytical methods. Biological evaluation revealed that compounds 4c and 4b showed the most potent antioxidant activity, with lower IC50 values than the standard, indicating strong free radical scavenging potential. In the cytotoxicity study against the MCF-7 breast cancer cell line, compound 4c exhibited the highest anticancer activity, followed by 4b, demonstrating a significant reduction in cell viability even at low concentrations. Molecular docking studies further supported these findings, where compound 4c revealed superior binding affinity compared to the reference drug Letrozole. Overall, the results highlight compound 4c as the most promising candidate with enhanced biological activity, suggesting its potential for further development as an effective anti-breast cancer agent, pending additional in vitro and in vivo validation.
REFERENCES


Eligros Kujur, Laxmi Banjare*, Design, Synthesis, And Anticancer Evaluation Of Novel Triazole Analogues As Aromatase Inhibitors For Breast Cancer, Int. J. Sci. R. Tech., 2026, 3 (6), 947-956. https://doi.org/10.5281/zenodo.20716941
 10.5281/zenodo.20716941
10.5281/zenodo.20716941