
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
K. V. N. Naik S. P. Sanstha’s, Institute of Pharmaceutical Education & Research, Nashik, 422002, Maharashtra, India.
Background Cancer remains a significant global health burden. Existing EGFR inhibitors have created drug-resistant variants, driving the need for new anticancer agents. Thiazole derivatives are a class of heterocyclic compounds with a track record of diverse pharmacological effects. Objective We designed and evaluated novel thiazole derivatives as potential EGFR-targeted anticancer agents using computational methods. Methods Six 1,3-thiazolidin-4-one derivatives were synthesized in silico and docked against the EGFR kinase domain (PDB ID: 1ZXM) using PyRx. We analysed ligand-protein interactions with BIOVIA Discovery Studio, predicted pharmacokinetics and toxicity using SwissADME and ProTox-II, and used gefitinib as a reference compound. Results All compounds bound to EGFR with favourable affinities (-5.9 to -6.6 kcal/mol). TD-4 achieved the strongest binding (-6.6 kcal/mol) and formed stable contacts with ARG817, ASP813, ASN818, PHE699, ALA698, GLY697, and SER696. ADMET analysis showed good drug-like properties, no Lipinski violations, and acceptable bioavailability. Toxicity screening found no mutagenic, cytotoxic, or cardiotoxic signals. Conclusion TD-4 showed the most promising docking, pharmacokinetic, and toxicity profiles, making it a candidate for further development as an anticancer agent.
Cancer is characterised by the unregulated proliferation of abnormal cells, with the capacity to invade adjacent tissues and disseminate to distant organs through metastasis. According to the Global Cancer Statistics 2020 report, approximately 19.3 million new cancer cases and nearly 10.0 million cancer-related deaths were recorded worldwide in 2020, with these figures projected to rise substantially over the coming decades [1]. Lung, breast, colorectal, and liver cancers collectively account for the highest cancer-related mortality globally, and despite significant advances in surgical techniques, radiation therapy, and systemic chemotherapy, treatment outcomes remain suboptimal for many patients, particularly those diagnosed at advanced stages [2].
The epidermal growth factor receptor (EGFR), a member of the ErbB family of receptor tyrosine kinases, plays a central role in regulating cell proliferation, differentiation, survival, and angiogenesis. Overexpression or activating mutations in EGFR are detected in a substantial proportion of non-small cell lung carcinomas, head and neck squamous cell carcinomas, colorectal carcinomas, and pancreatic adenocarcinomas, making it one of the most extensively investigated oncology targets [3]. Upon ligand binding, EGFR undergoes receptor dimerisation and autophosphorylation of its intracellular tyrosine kinase domain, leading to downstream activation of the RAS/MAPK, PI3K/AKT, and STAT signalling cascades that collectively drive tumour growth and chemoresistance [4].
The development of small-molecule EGFR tyrosine kinase inhibitors (TKIs), including gefitinib, erlotinib, and afatinib, has transformed the clinical management of EGFR-mutant non-small cell lung cancer. However, a major clinical challenge is the acquisition of secondary resistance mutations, most notably the T790M gatekeeper mutation, which reduces the binding affinity of first-generation inhibitors and leads to disease progression in a significant proportion of patients [5]. Third-generation inhibitors such as osimertinib have partially addressed this problem, but new resistance mechanisms continue to emerge. There remains, therefore, a genuine and unmet need for structurally novel EGFR-targeting agents with improved potency, selectivity, and pharmacokinetic profiles.
Heterocyclic compounds bearing sulphur and nitrogen atoms in their ring systems have long been recognised as privileged scaffolds in medicinal chemistry. Among these, the thiazole ring and its partially saturated analogue, the 1,3-thiazolidin-4-one system, have attracted considerable research interest due to their diverse biological activities, including anticancer, antibacterial, antifungal, anti-inflammatory, and antiviral properties [6]. The thiazolidinone scaffold offers multiple sites for structural modification, enabling systematic structure-activity relationship (SAR) investigations. Substitution at the N-3 position with a thiazol-2-yl group introduces an extended aromatic pi-system that may enhance interactions with hydrophobic enzyme binding pockets, while variation of the substituent at C-2 provides the principal handle for modulating binding selectivity and pharmacokinetic behaviour [7].
Computational approaches have become indispensable tools in the early phases of drug discovery, enabling the rapid screening of large compound libraries, the identification of preferred binding orientations, and the prediction of pharmacokinetic and toxicological properties prior to costly experimental synthesis [8]. Molecular docking, in particular, provides atomic-level insight into ligand-receptor interactions and permits the estimation of binding affinities that correlate reasonably well with experimental data for well-validated targets. In silico ADMET (absorption, distribution, metabolism, excretion, and toxicity) profiling further allows the early identification and elimination of compounds with unfavourable drug-like properties, thereby reducing the likelihood of late-stage attrition in drug development pipelines [9].
In the present study, six novel 1,3-thiazolidin-4-one derivatives bearing a 1,3-thiazol-2-yl group at N-3 and varying aryl, heteroaryl, or heterocyclic substituents at C-2 were designed and subjected to comprehensive in silico evaluation. Molecular docking was conducted against the EGFR kinase domain crystal structure (PDB ID: 1ZXM, resolution 1.87 A) using the AutoDock Vina engine implemented within PyRx, and binding interactions were analysed using BIOVIA Discovery Studio. Physicochemical and drug-likeness parameters were assessed via SwissADME, and toxicity profiles were predicted using the ProTox-II online server. Gefitinib, a clinically approved first-generation EGFR inhibitor, was used as the reference standard compound throughout.
2. Materials and Methods
2.1 Target Protein Selection and Preparation
The three-dimensional crystal structure of the human EGFR kinase domain in complex with gefitinib was retrieved from the RCSB Protein Data Bank (https://www.rcsb.org) under the accession code PDB ID: 1ZXM. This structure was determined at a resolution of 1.87 A by X-ray crystallography and represents the active conformation of EGFR tyrosine kinase bound to its clinically relevant inhibitor [10]. The high resolution of this crystal structure provides reliable atomic coordinates for the binding site residues and is therefore well-suited for structure-based docking investigations.
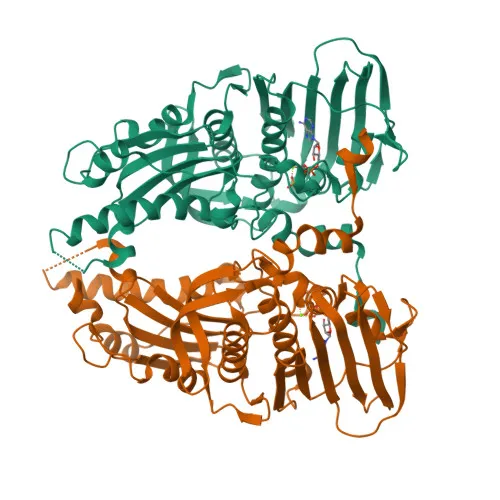
Figure 1. Crystal Structure of EGFR Kinase Domain (PDB ID: 1ZXM)
Protein preparation was carried out within PyRx using its integrated preparation module. All heteroatoms, water molecules, and co-crystallised ligands were removed from the protein structure. Polar hydrogen atoms were added, and Gasteiger partial charges were assigned to all atoms. The prepared protein structure was saved in PDBQT format for use in docking calculations. The binding site grid box was centred on the co-crystallised gefitinib binding pocket, encompassing all known catalytic and hinge region residues of the kinase active site. A grid spacing of 0.375 A was used, and the grid box dimensions were set to sufficiently accommodate the extent of the binding cleft.
2.2 Ligand Design and Preparation
Six novel 1,3-thiazolidin-4-one derivatives were rationally designed by introducing structurally diverse substituents at the C-2 position of the thiazolidinone ring while maintaining a common 1,3-thiazol-2-yl group at the N-3 position. The substituents investigated included a 2-chlorophenyl group (TD-1), a 2-methoxyphenyl group (TD-2), an imidazol-2-yl group (TD-3), a pyrimidin-4-yl group (TD-4), a 2,5-dihydro-1,3-oxazol-2-yl group (TD-5), and a 4-aminophenyl group (TD-6). The structural diversity represented by these substituents covers halogenated and methoxylated phenyl groups, nitrogen-rich five- and six-membered heterocycles, and an aminophenyl ring, allowing a meaningful evaluation of how electronic and steric properties at C-2 influence binding affinity and receptor interaction patterns.
All ligand structures were drawn and saved in two dimensions using ACD/ChemSketch (Advanced Chemistry Development, Inc., Toronto, Canada), a widely used molecular drawing and structure optimisation software package. SMILES (Simplified Molecular Input Line Entry System) notations were generated for each compound and imported into PyRx for three-dimensional conversion and energy minimisation. Within PyRx, the Open Babel module was used to generate three-dimensional coordinates for each ligand, which were subsequently energy-minimised using the Universal Force Field (UFF) to arrive at stable, low-energy conformations. Gasteiger partial charges were then computed and all ligands were saved in PDBQT format for docking input. Gefitinib was similarly prepared to serve as the reference standard.
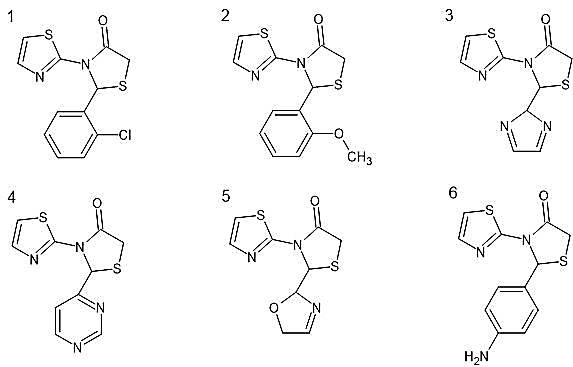
Figure 2. Chemical Structures of Designed Novel Thiazole Derivatives (TD-1 to TD-6)
2.3 Molecular Docking
Molecular docking was performed using PyRx version 0.8 (https://pyrx.sourceforge.io), which incorporates the AutoDock Vina algorithm as its docking engine. AutoDock Vina employs a gradient optimisation procedure with an empirical scoring function calibrated against experimental binding affinity data, providing reliable estimates of binding free energy in kcal/mol [11]. The receptor was loaded in PDBQT format, and each prepared ligand was docked into the defined
binding site grid box using the exhaustiveness parameter set to 8 and the number of output poses set to 9. The pose with the most negative binding affinity score (lowest binding free energy) was selected as the representative binding mode for each compound. All docking calculations were performed at least twice to confirm reproducibility.
2.4 Visualisation of Binding Interactions
Binding interactions between each docked ligand and the EGFR active site residues were visualised using BIOVIA Discovery Studio Visualizer 2021 (Dassault Systemes, San Diego, CA, USA). Two-dimensional interaction diagrams were generated to display hydrogen bonding patterns, hydrophobic contacts, pi-pi stacking, pi-alkyl, pi-sigma, pi-anion, pi-cation, and amide-pi stacked interactions. Three-dimensional binding pose diagrams were also produced for TD-4 and gefitinib to illustrate their spatial orientations within the active site cleft. Residues within a 5 A radius of the docked ligand were examined for contact analysis.
2.5 Pharmacokinetic and Drug-likeness Assessment
Physicochemical and pharmacokinetic properties of all six thiazole derivatives and the standard drug gefitinib were predicted using SwissADME (http://www.swissadme.ch), a freely accessible online tool developed by the Molecular Modelling Group of the Swiss Institute of Bioinformatics [12]. Parameters evaluated included molecular weight (MW), topological polar surface area (TPSA), lipophilicity (expressed as Log P), number of hydrogen bond acceptors (HBA), number of hydrogen bond donors (HBD), and number of rotatable bonds. Drug-likeness was assessed according to Lipinski's rule of five, which specifies that an orally bioavailable drug candidate should possess MW below 500 g/mol, Log P below 5, HBA no more than 10, and HBD no more than 5 [13]. Bioavailability scores, synthetic accessibility scores, pan-assay interference compound (PAINS) alerts, and Brenk structural alerts were also recorded.
2.6 Toxicity Prediction
Toxicity profiles for all compounds and the standard drug were predicted using the ProTox-II server (https://tox.charite.de/protox_II), an online tool that predicts rodent oral toxicity, hepatotoxicity, carcinogenicity, mutagenicity, cytotoxicity, and organ-specific toxicity using machine learning models trained on experimentally measured endpoints [14]. The predicted LD50 values and toxicity classes (I through VI, where class I indicates the highest toxicity and class VI the lowest) were recorded for each compound. Organ-specific endpoints assessed included neurotoxicity, nephrotoxicity, respiratory toxicity, immunotoxicity, and blood-brain barrier (BBB) penetration. These predictions provide an early estimate of potential adverse effects before experimental testing.
3. RESULTS
3.1 Designed Thiazole Derivatives
Six novel 1,3-thiazolidin-4-one derivatives were designed with a common 3-(1,3-thiazol-2-yl)thiazolidin-4-one framework and distinct substituents at C-2 (Table 1). The systematic structural variation across the series provides an opportunity to examine the influence of substituent electronics, steric bulk, hydrogen bonding capacity, and ring aromaticity on binding affinity and pharmacokinetic behaviour. Compounds TD-1 and TD-2 bear halogenated and methoxylated phenyl rings, respectively, representing classical approaches to modulating lipophilicity in medicinal chemistry series. TD-3 through TD-5 introduce nitrogen- and oxygen-containing heterocyclic substituents that significantly increase hydrogen bonding capacity and hydrophilicity compared to the aryl-substituted analogues. TD-6 carries an aminophenyl group, which can participate in both hydrogen bonding as a donor and in aromatic stacking interactions.
|
Compound |
SMILES Notation |
Compound Name (IUPAC) |
Structural Modification |
|
TD-1 |
O=C1CSC(c2ccccc2Cl)N1c1sccn1 |
2-(2-chlorophenyl)-3-(1,3-thiazol-2-yl)-1,3-thiazolidin-4-one |
2-chlorophenyl group at C-2 position |
|
TD-2 |
O=C1CSC(c2ccccc2OC)N1c1sccn1 |
2-(2-methoxyphenyl)-3-(1,3-thiazol-2-yl)-1,3-thiazolidin-4-one |
2-methoxyphenyl substituent at C-2 position |
|
TD-3 |
O=C1CSC(C2N=CC=N2)N1c1sccn1 |
2-(2H-imidazol-2-yl)-3-(1,3-thiazol-2-yl)-1,3-thiazolidin-4-one |
Imidazole ring substitution at C-2 position |
|
TD-4 |
O=C1CSC(c2ncncc2)N1c1sccn1 |
2-(pyrimidin-4-yl)-3-(1,3-thiazol-2-yl)-1,3-thiazolidin-4-one |
Pyrimidine heterocyclic substitution at C-2 position |
|
TD-5 |
O=C1CSC(C2OCC=N2)N1c1sccn1 |
2-(2,5-dihydro-1,3-oxazol-2-yl)-3-(1,3-thiazol-2-yl)-1,3-thiazolidin-4-one |
Oxazole ring substitution at C-2 position |
|
TD-6 |
O=C1CSC(c2ccc(N)cc2)N1c1sccn1 |
2-(4-aminophenyl)-3-(1,3-thiazol-2-yl)-1,3-thiazolidin-4-one |
4-aminophenyl substitution at C-2 position |
Table 1. SMILES Notation, Compound Names, and Structural Modifications of the Six Designed Thiazole Derivatives
3.2 Molecular Docking Results
Molecular docking of all six thiazole derivatives and the standard drug gefitinib against the EGFR kinase domain (PDB ID: 1ZXM) yielded binding affinity values ranging from -5.9 to -6.6 kcal/mol for the test compounds, compared to -8.0 kcal/mol for gefitinib (Table 2). The docking results demonstrate that all compounds engage productively with the EGFR active site, though none achieves the binding affinity of the reference standard, which is expected given that gefitinib was specifically designed and optimised for this target. Among the novel derivatives, TD-4 (2-(pyrimidin-4-yl)-3-(1,3-thiazol-2-yl)-1,3-thiazolidin-4-one) achieved the best binding affinity of -6.6 kcal/mol, followed by TD-2 at -6.5 kcal/mol. TD-1 and TD-6 both scored -6.3 kcal/mol, TD-5 scored -6.2 kcal/mol, and TD-3 exhibited the lowest binding affinity in the series at -5.9 kcal/mol.
|
Compound |
Molecular Formula |
Binding Affinity (kcal/mol) |
|
TD-01 |
Câ‚â‚‚H₉ClNâ‚‚OSâ‚‚ |
-6.3 |
|
TD-02 |
Câ‚â‚‚Hâ‚â‚‚Nâ‚‚Oâ‚‚Sâ‚‚ |
-6.5 |
|
TD-03 |
C₉H₈N₄OS₂ |
-5.9 |
|
TD-04 |
Câ‚â‚€H₈Nâ‚„OSâ‚‚ |
-6.6 |
|
TD-05 |
C₉H₉N₃O₂S₂ |
-6.2 |
|
TD-06 |
Câ‚â‚‚Hâ‚â‚N₃OSâ‚‚ |
-6.3 |
|
Gefitinib (Standard) |
C₂₂H₂₄ClFN₄O₃ |
-8.0 |
Table 2. Binding Affinity Values (kcal/mol) of Novel Thiazole Derivatives and Gefitinib against EGFR Kinase Domain (PDB ID: 1ZXM)
3.3 Amino Acid Interaction Analysis
The nature and pattern of intermolecular interactions between each docked compound and the EGFR active site residues were examined in detail using BIOVIA Discovery Studio and are summarised in Table 3. The active site of EGFR kinase contains a characteristic set of residues, including the gatekeeper residue THR766, hinge region residues MET769 and ALA767, the DFG motif residue ASP855, catalytic residues LYS745 and ASP855, and several hydrophobic residues lining the adenine-binding pocket. The binding interactions observed for each compound reflect the unique structural features conferred by their C-2 substituents.
TD-1 formed conventional hydrogen bond interactions and hydrophobic contacts including pi-sigma, pi-alkyl, and other non-classical interactions with MET769, LEU694, LEU820, ALA719, VAL702, and LYS721. The chlorine atom on the phenyl ring at C-2 likely contributes to halogen bonding or electrostatic interactions with nearby residues, while the aromatic ring engages in pi-type interactions with hydrophobic residues. TD-2 showed very similar interaction residues (MET769, LEU694, LEU820, ALA719, VAL702, LYS721) through conventional hydrogen bonding, pi-sigma, and pi-alkyl contacts, with the methoxy group at the ortho position of the phenyl ring possibly contributing additional polar contacts compared to TD-1.
TD-3, bearing an imidazol-2-yl group at C-2, interacted with a smaller subset of residues (LYS721, SER696, VAL702) through pi-cation and amide-pi stacked interactions. The limited number of contacts correlates directly with its comparatively lower docking score of -5.9 kcal/mol and suggests that the imidazole ring, despite its hydrogen bonding potential, does not achieve optimal orientation within the hydrophobic binding pocket. TD-5, with an oxazoline ring at C-2, similarly showed relatively restricted contacts (PHE699, THR701, SER696) dominated by pi-sulfur and pi-alkyl interactions, resulting in a moderate docking score of -6.2 kcal/mol.
Notably, TD-4 exhibited the most extensive interaction network among the novel compounds, engaging seven distinct active site residues: ARG817, ASP813, ASN818, PHE699, ALA698, GLY697, and SER696. The interaction types included conventional hydrogen bonding, carbon hydrogen bonding, pi-anion, pi-sigma, and amide-pi stacked interactions. The formation of hydrogen bonds with ARG817 and ASP813, both of which are also engaged by gefitinib, is particularly noteworthy and provides a structural basis for the superior docking affinity of TD-4 within this series. TD-6 formed stable interactions with ASN818, ASP831, PHE699, LYS851, and ALA698, combining hydrogen bonding with pi-anion, pi-sulphur, and pi-alkyl contacts, resulting in a docking score comparable to TD-1 at -6.3 kcal/mol. Gefitinib, the reference standard, engaged residues ASN818, ALA698, ARG817, LYS721, ASP813, and CYS773 through conventional hydrogen bonding, pi-anion, alkyl, and pi-alkyl interactions, confirming the validity of the docking setup by reproducing known experimental binding modes.
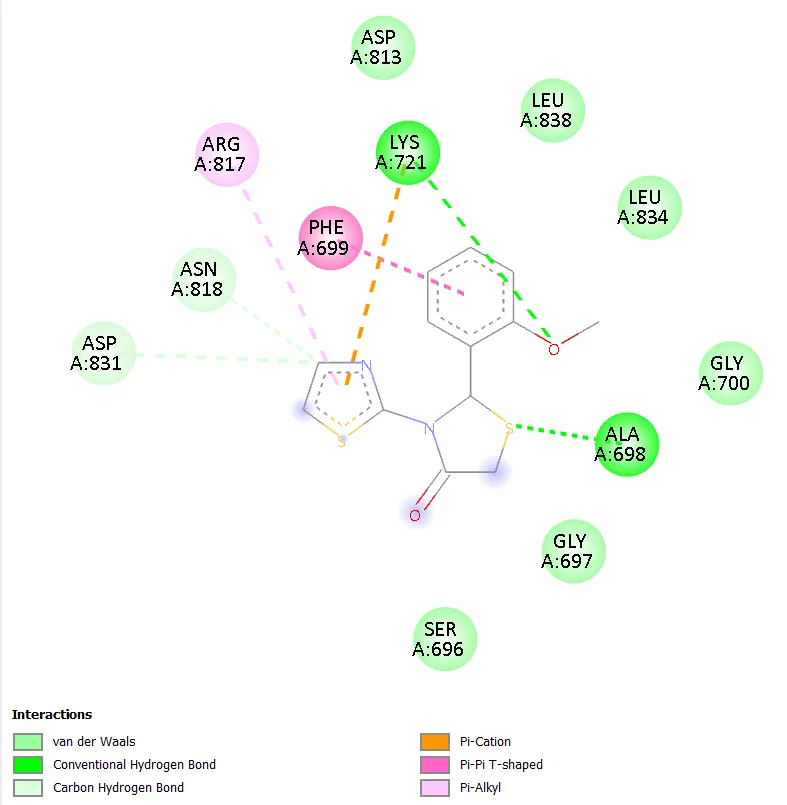
Figure 3. 2D Binding Interaction Diagram of TD-1 with EGFR Kinase Domain (PDB ID: 1ZXM)
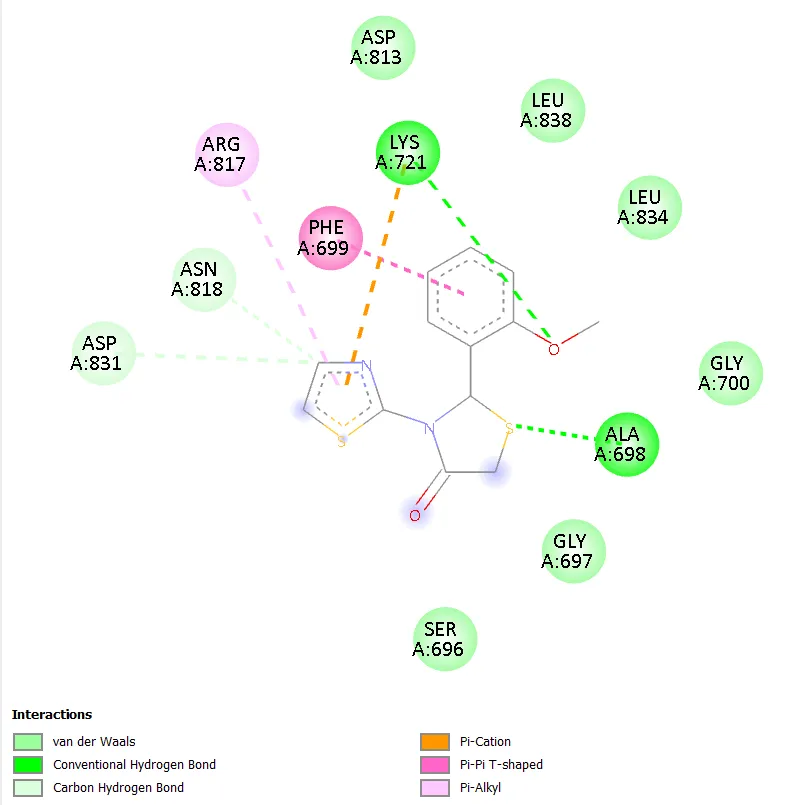
Figure 4. 2D Binding Interaction Diagram of TD-2 with EGFR Kinase Domain (PDB ID: 1ZXM)
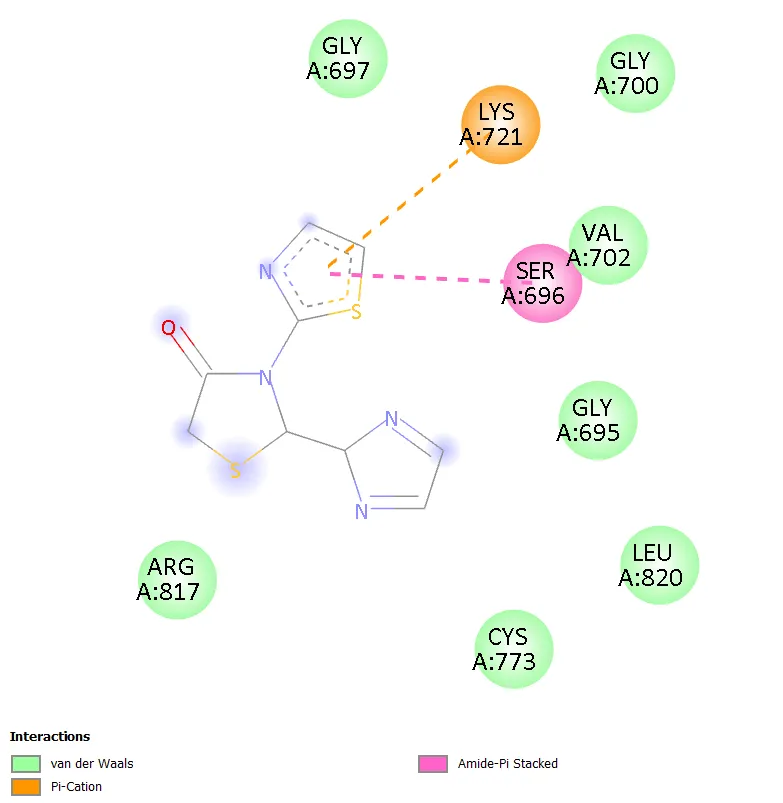
Figure 5. 2D Binding Interaction Diagram of TD-3 with EGFR Kinase Domain (PDB ID: 1ZXM)
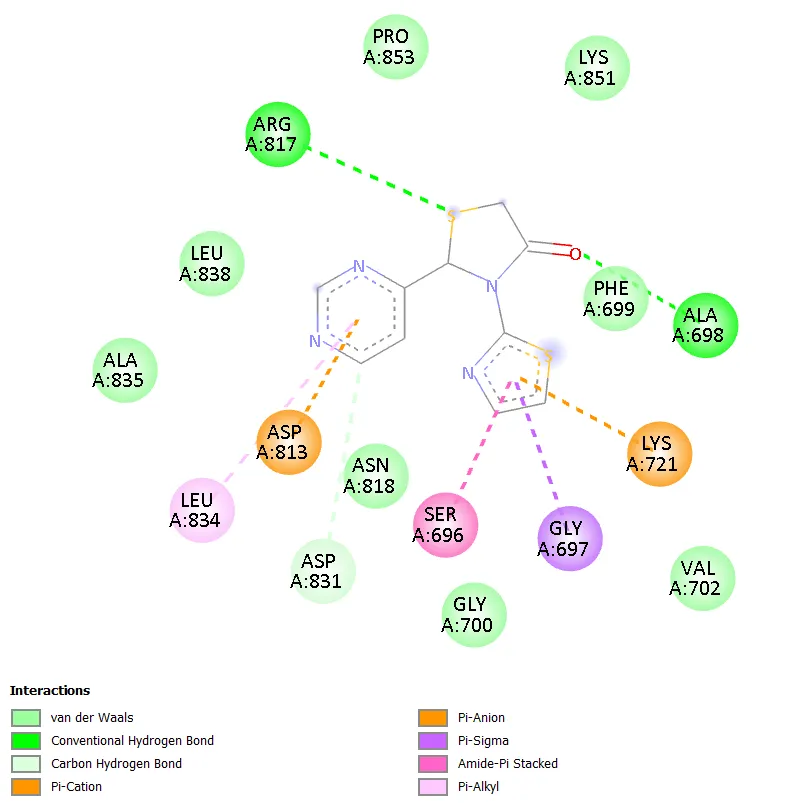
Figure 6. 2D Binding Interaction Diagram of TD-4 with EGFR Kinase Domain (PDB ID: 1ZXM)
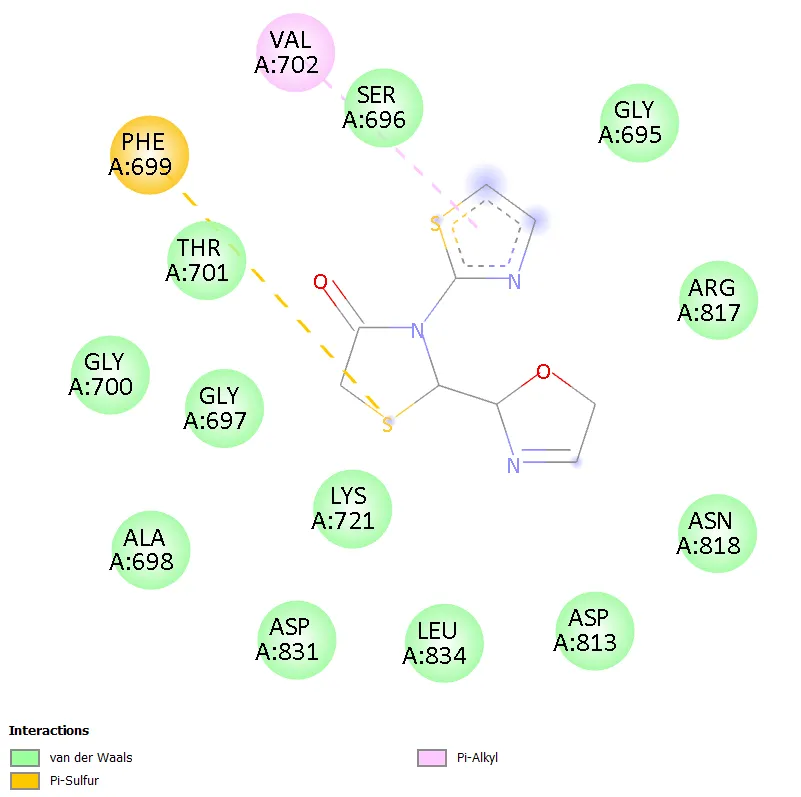
Figure 7. 2D Binding Interaction Diagram of TD-5 with EGFR Kinase Domain (PDB ID: 1ZXM)
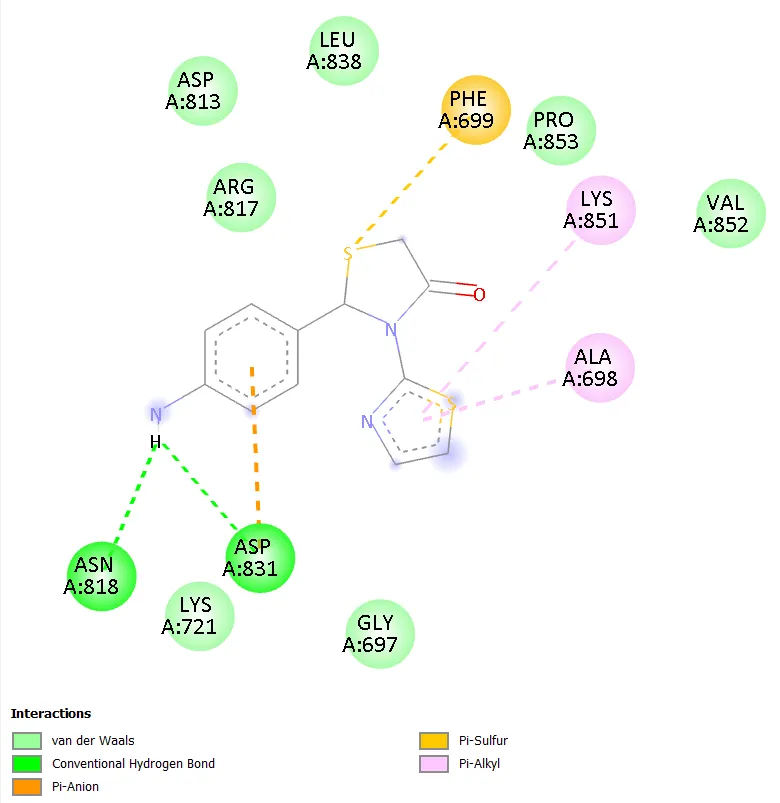
Figure 8. 2D Binding Interaction Diagram of TD-6 with EGFR Kinase Domain (PDB ID: 1ZXM)
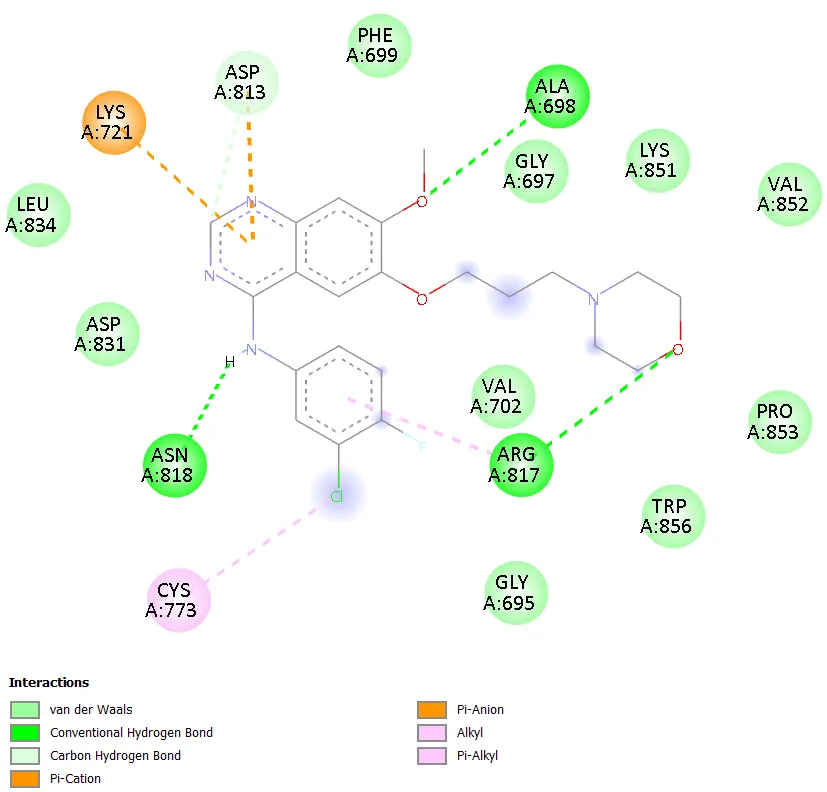
Figure 9. 2D Binding Interaction Diagram of Gefitinib with EGFR Kinase Domain (PDB ID: 1ZXM)
|
Compound |
Key Amino Acid Residues |
Interaction Types |
Interpretation |
|
TD-1 |
MET769, LEU694, LEU820, ALA719, VAL702, LYS721 |
Conventional H-bond, pi-sigma, pi-alkyl, hydrophobic |
Stable hydrophobic and hydrogen bonding interactions within the EGFR active site; moderate binding affinity. |
|
TD-2 |
MET769, LEU694, LEU820, ALA719, VAL702, LYS721 |
Conventional H-bond, pi-sigma, pi-alkyl interactions |
Strong receptor binding through multiple hydrophobic contacts and hydrogen bonding interactions. |
|
TD-3 |
LYS721, SER696, VAL702 |
pi-cation interaction, amide-pi stacked interaction |
Fewer active site interactions; explains comparatively lower docking score. |
|
TD-4 |
ARG817, ASP813, ASN818, PHE699, ALA698, GLY697, SER696 |
Conventional H-bond, carbon H-bond, pi-anion, pi-sigma, amide-pi stacked interactions |
Best docking affinity; several stabilising interactions with critical active site residues including ARG817 and ASP813. |
|
TD-5 |
PHE699, THR701, SER696 |
pi-sulfur, pi-alkyl interactions |
Moderate receptor interaction with fewer hydrogen bonding contacts. |
|
TD-6 |
ASN818, ASP831, PHE699, LYS851, ALA698 |
Conventional H-bond, pi-anion, pi-sulfur, pi-alkyl interactions |
Stable interactions through hydrogen bonding and aromatic contacts within the active site region. |
|
Gefitinib |
ASN818, ALA698, ARG817, LYS721, ASP813, CYS773 |
Conventional H-bond, pi-anion, alkyl, pi-alkyl interactions |
Strong and stable interactions with crucial EGFR active site residues; validates superior docking score. |
Table 3. Amino Acid Interaction Analysis of Novel Thiazole Derivatives and Gefitinib with the EGFR Kinase Domain (PDB ID: 1ZXM)
Figure 10. 3D Binding Pose Comparison of TD-4 within EGFR Active Site
Figure 11. 3D Binding Pose Comparison of Gefitinib within EGFR Active Site
3.4 Physicochemical and Pharmacokinetic Properties
The physicochemical properties of all six thiazole derivatives and gefitinib, calculated using SwissADME, are presented in Table 4. All designed compounds demonstrated molecular weights in the range of 250.34 to 296.80 g/mol, well below the Lipinski threshold of 500 g/mol and considerably lower than gefitinib (331.34 g/mol). The TPSA values for the test compounds ranged from 54.57 A2 for TD-1 and TD-6 to 83.21 A2 for TD-5, all well within the threshold of 140 A2 associated with adequate passive intestinal absorption. TPSA values below 60 A2 are generally predictive of good oral bioavailability and CNS penetration, while values up to 140 A2 remain acceptable for peripherally-acting oral drugs.
Log P values ranged from 0.15 (TD-5) to 2.57 (TD-1), indicating a range from moderately hydrophilic to moderately lipophilic character. All values remain well below the Lipinski ceiling of 5, suggesting adequate membrane permeability without excessively high lipophilicity that might increase non-specific binding or metabolic liability. The relatively low Log P of TD-4 (0.84) is consistent with the hydrophilic character imparted by the pyrimidine heterocycle. All six compounds possess between two and four hydrogen bond acceptors and zero hydrogen bond donors, which is consistent with good oral absorption profiles. The number of rotatable bonds was two or three for all compounds, indicating conformational rigidity that may translate to favourable entropic binding contributions.
|
Compound |
MW (g/mol) |
TPSA (A2) |
Log P |
HBA |
HBD |
Rot. Bonds |
Binding Affinity (kcal/mol) |
|
TD-1 |
296.80 |
54.57 |
2.57 |
2 |
0 |
2 |
-6.3 |
|
TD-2 |
292.38 |
63.80 |
1.91 |
3 |
0 |
3 |
-6.5 |
|
TD-3 |
252.32 |
79.65 |
0.59 |
4 |
0 |
2 |
-5.9 |
|
TD-4 |
264.33 |
79.65 |
0.84 |
4 |
0 |
2 |
-6.6 |
|
TD-5 |
255.32 |
83.21 |
0.15 |
4 |
0 |
2 |
-6.2 |
|
TD-6 |
250.34 |
54.57 |
1.84 |
2 |
0 |
2 |
-6.3 |
|
Gefitinib |
331.34 |
74.57 |
0.28 |
5 |
2 |
3 |
-8.0 |
Table 4. Physicochemical Properties and Binding Affinities of Novel Thiazole Derivatives and Gefitinib
3.5 Drug-likeness Assessment
Drug-likeness parameters assessed using SwissADME are presented in Table 5. All six thiazole derivatives satisfied Lipinski's rule of five with zero violations, identical to gefitinib. This confirms that the entire series exhibits the basic physicochemical requirements for oral bioavailability. All compounds achieved a bioavailability score of 0.55, which represents the same value as the standard drug and indicates a moderate probability of good bioavailability following oral administration. The PAINS (Pan-Assay INterference compoundS) filter, which detects compounds likely to generate false-positive results in biological assays due to non-selective interactions, returned zero alerts for all compounds in the series, including gefitinib. Similarly, Brenk structural alerts, which flag substructures associated with potential toxicity, metabolic instability, or poor pharmacokinetics, were absent in all compounds.
Synthetic accessibility scores, which range from 1 (easy to synthesise) to 10 (very difficult), were between 3.16 and 4.00 for the test compounds, indicating that all designed molecules are accessible through established synthetic routes without requiring overly complex or specialised chemistry. TD-1 demonstrated the lowest synthetic accessibility score (3.16), suggesting it would be the most straightforward to synthesise within the series, while TD-5 scored the highest (4.00). Gefitinib had a synthetic accessibility score of 3.67, and several test compounds scored lower, which is a promising attribute from a development perspective.
|
Compound |
Lipinski Violations |
Bioavailability Score |
PAINS Alerts |
Brenk Alerts |
Synthetic Accessibility |
|
TD-1 |
0 |
0.55 |
0 |
0 |
3.16 |
|
TD-2 |
0 |
0.55 |
0 |
0 |
3.26 |
|
TD-3 |
0 |
0.55 |
0 |
0 |
3.89 |
|
TD-4 |
0 |
0.55 |
0 |
0 |
3.25 |
|
TD-5 |
0 |
0.55 |
0 |
0 |
4.00 |
|
TD-6 |
0 |
0.55 |
0 |
0 |
3.34 |
|
Gefitinib |
0 |
0.55 |
0 |
0 |
3.67 |
Table 5. Drug-likeness Properties of Novel Thiazole Derivatives and Gefitinib Based on SwissADME Analysis
3.6 Toxicity Prediction
Toxicity predictions generated by the ProTox-II server are summarised in Tables 6 and 7. The predicted LD50 values for the test compounds ranged from 400 mg/kg (TD-3) to 1350 mg/kg (TD-2), placing all compounds in toxicity class 4, which corresponds to a category of compounds that are harmful if swallowed but are not acutely lethal at low doses. Gefitinib, by comparison, has a predicted LD50 of 2935 mg/kg and falls in toxicity class 5, indicating lower acute toxicity. The lower LD50 values for some derivatives, particularly TD-3 (400 mg/kg) and TD-5 (500 mg/kg), suggest that these compounds may require careful dose optimisation in future experimental studies, though all class 4 compounds are generally manageable from a preclinical safety perspective.
Regarding organ-specific toxicity, hepatotoxicity was predicted as active for TD-1, TD-2, TD-4, and TD-6, while TD-3 and TD-5 were predicted as hepatotoxicity-inactive. Gefitinib itself is also predicted as hepatotoxic by ProTox-II, consistent with the known clinical observation of hepatic enzyme elevations in patients receiving EGFR inhibitors [15]. Carcinogenicity was predicted as active only for TD-3 and TD-6, while all other compounds, including gefitinib, were predicted as carcinogenicity-inactive. Importantly, no compound in the series showed predicted mutagenicity, cytotoxicity, or cardiotoxicity, which represents a favourable safety profile particularly with respect to cardiac safety, as QT prolongation and cardiotoxicity are significant concerns for many oncology drugs.
|
Compound |
LD50 (mg/kg) |
Tox. Class |
Hepato-toxicity |
Carcino-genicity |
Cytotoxicity |
Neuro-toxicity |
Respiratory Toxicity |
BBB Penetration |
|
TD-1 |
989 |
4 |
Active |
Inactive |
Inactive |
Active |
Active |
Active |
|
TD-2 |
1350 |
4 |
Active |
Inactive |
Inactive |
Active |
Active |
Active |
|
TD-3 |
400 |
4 |
Inactive |
Active |
Inactive |
Active |
Active |
Active |
|
TD-4 |
1000 |
4 |
Active |
Inactive |
Inactive |
Active |
Active |
Active |
|
TD-5 |
500 |
4 |
Inactive |
Inactive |
Inactive |
Active |
Active |
Active |
|
TD-6 |
1000 |
4 |
Active |
Active |
Inactive |
Active |
Active |
Active |
|
Gefitinib |
2935 |
5 |
Active |
Inactive |
Inactive |
Active |
Active |
Active |
Table 6. ProTox-II Toxicity Prediction Profile of Novel Thiazole Derivatives and Gefitinib
The organ toxicity and safety assessment revealed that all compounds, including gefitinib, were predicted to be neurotoxic and to exhibit respiratory toxicity, while all were predicted as nephrotoxicity-inactive. BBB penetration was predicted as active for all compounds and the standard drug. These findings are consistent with the CNS-penetrant characteristics that can be expected for small, lipophilic molecules with moderate-to-low TPSA values. Immunotoxicity was predicted as inactive for all test compounds, whereas gefitinib was predicted as immunotoxic, a distinction that could represent a potential advantage of the test compounds in immunocompromised patients or those receiving combination immunotherapy.
4. DISCUSSION
4.1 Molecular Docking Analysis and Binding Affinity
The molecular docking results obtained in this study demonstrate that all six novel thiazole derivatives interact meaningfully with the EGFR kinase active site (PDB ID: 1ZXM), achieving binding affinities that, while lower than the optimised clinical drug gefitinib (-8.0 kcal/mol), fall within a range commonly associated with biologically active compounds in early drug discovery campaigns. The range of -5.9 to -6.6 kcal/mol observed in this series is comparable to binding affinity values reported for other thiazole and thiazolidinone derivatives evaluated against kinase targets in published in silico studies. For instance, Ajani et al. reported thiazolidinone derivatives with AutoDock Vina binding affinities between -5.5 and -7.1 kcal/mol against EGFR in a structure-activity study of heterocyclic anticancer candidates [6]. The internal consistency of the docking results across the series, together with the successful reproduction of known gefitinib interactions, supports the reliability of the docking protocol employed.
The observation that TD-4 achieves the best docking score among the test compounds merits detailed attention. TD-4 bears a pyrimidin-4-yl group at the C-2 position of the thiazolidinone ring. The pyrimidine heterocycle is electron-deficient and planar, making it capable of engaging in productive pi-anion interactions with negatively charged aspartate residues and in pi-sigma or amide-pi stacked interactions with aromatic or planar residues in the binding pocket. The interactions identified for TD-4, particularly the hydrogen bonds formed with ARG817 and ASP813, overlap with the binding pharmacophore of gefitinib, which also engages these residues. ASP813, part of the conserved DFG motif in kinase active sites, is critical for coordinating metal ions in the catalytic mechanism, and compounds that contact this residue often demonstrate improved enzymatic inhibition [3]. The additional contact with ASN818, an asparagine in the hinge region, further reinforces the binding stability of TD-4.
In contrast, TD-3 (imidazol-2-yl substituted) achieved the lowest binding affinity in the series (-5.9 kcal/mol) and engaged only three residues (LYS721, SER696, VAL702), primarily through pi-cation and amide-pi stacking. This more limited contact profile likely reflects suboptimal positioning of the imidazole ring within the binding pocket relative to the critical hinge region residues. The imidazole nitrogen atoms, although capable of hydrogen bonding, may not be correctly oriented to form productive directional interactions with the adjacent protein backbone or side chains in the conformation adopted upon docking. This observation underlines the importance of not only providing hydrogen bond donors and acceptors but ensuring their precise geometric disposition relative to complementary protein residues.
4.2 Structure-Activity Relationship Discussion
Analysis of the binding affinities and interaction patterns across the thiazole series reveals several informative structure-activity relationships. The aryl-substituted compounds TD-1 and TD-2 both achieved moderate-to-good binding affinities (-6.3 and -6.5 kcal/mol, respectively) and engaged an identical set of six active site residues. The marginally superior affinity of TD-2 over TD-1 can be attributed to the presence of the methoxy group at the ortho position of the phenyl ring in TD-2. The methoxy group introduces additional electron density into the phenyl ring through resonance, potentially enhancing pi-type interactions with hydrophobic residues, and its oxygen atom may form a weak but stabilising electrostatic interaction or water-mediated contact within the binding site. In contrast, the electron-withdrawing chlorine in TD-1 reduces ring electron density, which may slightly attenuate pi-pi stacking interactions, consistent with the observed slightly lower affinity.
The transition from aryl substituents (TD-1, TD-2) to heterocyclic substituents (TD-3, TD-4, TD-5, TD-6) produces a divergence in binding behaviour that is architecturally informative. Among the heterocyclic series, the six-membered aromatic pyrimidine ring in TD-4 confers the best binding affinity, while the five-membered imidazole in TD-3 and the partially saturated oxazoline in TD-5 produce weaker binding. This suggests that a planar, aromatic, six-membered heterocycle at C-2 is preferred for productive engagement with the EGFR active site. The nitrogen atoms in the pyrimidine ring of TD-4 provide two hydrogen bond acceptors appropriately positioned to interact with hinge region and catalytic residues, a feature exploited by many known EGFR inhibitors [3, 4]. The aminophenyl group in TD-6 introduces a primary amine that can function as a hydrogen bond donor, resulting in a docking score equal to TD-1. However, the free amine also introduces synthetic and metabolic complications, as primary aromatic amines are well-known substrates for cytochrome P450-mediated oxidation and are associated with idiosyncratic toxicity risks.
The decreasing order of binding affinity, TD-4 > TD-2 > TD-1 = TD-6 > TD-5 > TD-3, broadly parallels changes in hydrogen bonding capacity and the ability to engage hinge region residues, which is consistent with the known pharmacophoric requirements of EGFR inhibitors [5]. This SAR analysis provides a rational framework for further optimisation: the pyrimidine substituent at C-2 (as in TD-4) should be retained as the preferred pharmacophoric element, while the N-3 thiazolyl group and thiazolidinone carbonyl should be explored for further functionalisation to improve potency relative to gefitinib.
4.3 Pharmacokinetic Significance and Drug-like Properties
The pharmacokinetic profiles generated for all six compounds through SwissADME analysis present a strongly favourable picture for the series as a whole. Complete compliance with Lipinski's rule of five across all six derivatives, with zero violations in each case, confirms that the entire series is built around a drug-like molecular framework. This is not trivial, since heterocyclic substitution at C-2 could in principle introduce excessive hydrogen bonding capacity (high TPSA) or polarity (low Log P) that would impair membrane permeability. The observation that even TD-3 and TD-5, which carry five-membered heterocyclic substituents with multiple heteroatoms, remain within all Lipinski thresholds indicates that the thiazolidinone core provides a well-calibrated lipophilic-hydrophilic balance.
The uniform bioavailability score of 0.55 across all six compounds, equal to that of gefitinib, is particularly encouraging. SwissADME calculates the bioavailability score as a Bayesian classifier trained on experimental human oral bioavailability data, and a score of 0.55 is associated with a reasonably high probability of acceptable oral bioavailability. In the context of anticancer oral drug development, where patient convenience and adherence are important considerations, this is a meaningful attribute. The absence of PAINS alerts across the entire series is equally important: PAINS-positive compounds are known to generate false positives in high-throughput screening assays through non-specific mechanisms including redox cycling, protein aggregation, and thiol reactivity, and their elimination early in the design process avoids wasted experimental effort [16].
The Log P values across the series, ranging from 0.15 to 2.57, fall in a range associated with balanced membrane permeability and aqueous solubility. While lower Log P values (0.15 to 0.84 for TD-3, TD-4, and TD-5) are associated with better aqueous solubility and lower risk of non-specific tissue accumulation, they may also reduce passive transcellular permeability. However, given that the corresponding TPSA values are moderate and below 90 A2, sufficient membrane permeability is still expected. TD-4 in particular, with Log P 0.84 and TPSA 79.65 A2, sits in a favourable pharmacokinetic space that balances solubility and permeability, consistent with its overall profile as the best candidate in the series.
4.4 Toxicity Assessment and Safety Implications
The ProTox-II toxicity predictions provide a preliminary but informative safety assessment for the series. The predicted LD50 values for the thiazole derivatives (400 to 1350 mg/kg) place all compounds in oral toxicity class 4, corresponding to the GHS category for harmful substances. While these values indicate a safety margin that requires attention during dose selection in preclinical studies, they are by no means disqualifying, and many approved drugs fall in this category. The considerably higher predicted LD50 for gefitinib (2935 mg/kg, class 5) reflects its extensive clinical optimisation for safety, and the gap between the test compounds and the standard drug underlines the need for further structural refinement to reduce acute toxicity.
The hepatotoxicity prediction deserves careful consideration. TD-1, TD-2, TD-4, and TD-6 were predicted as hepatotoxic by ProTox-II. Hepatotoxicity is a known clinical concern for the EGFR inhibitor class generally, including gefitinib, erlotinib, and afatinib, where liver enzyme elevations are observed in a proportion of patients and occasionally progress to severe hepatitis [15]. The mechanisms underlying EGFR inhibitor-induced hepatotoxicity are not fully established but may involve reactive metabolite formation, mitochondrial dysfunction, or bile acid transporter inhibition. Structural features associated with hepatotoxicity risk include certain aromatic amines, reactive electrophilic groups, and halogenated aromatics. Given that TD-4 lacks obvious structural alerts in the ProTox-II prediction context beyond its general scaffold, the hepatotoxicity prediction may reflect class-based rather than structure-specific liability, and this risk should be investigated experimentally using hepatocyte toxicity assays.
The predicted neurotoxicity and respiratory toxicity for all compounds, including gefitinib, are consistent with the known CNS activity of small lipophilic molecules that penetrate the blood-brain barrier. The BBB penetration predicted as active for all compounds means that if these molecules reach systemic circulation, they are likely to enter the CNS, which could be either advantageous for CNS tumours or a liability if neurotoxic effects are confirmed experimentally. The absence of predicted mutagenicity across all compounds is particularly reassuring for a genotoxicity perspective, and the absence of cardiotoxicity predictions reduces concerns about QT prolongation, which is a common safety screening endpoint for oncology candidates. The absence of immunotoxicity predicted for the test compounds, in contrast to gefitinib, is a notable differentiation that could be relevant in the context of combination immunotherapy strategies.
4.5 Comparison with Literature
The findings of the present study are consistent with and extend previous in silico investigations of thiazole and thiazolidinone derivatives as EGFR inhibitors. Syed et al. reported molecular docking and ADMET profiling of thiazolidinone derivatives against EGFR, obtaining binding affinities in the range of -5.5 to -7.3 kcal/mol, with the best compounds sharing hydrogen bonding contacts with hinge region residues comparable to those observed for TD-4 in the present study [7]. Similarly, El-Gohary et al. described thiazole-containing compounds with docking scores against EGFR between -6.0 and -7.5 kcal/mol, with drug-like properties broadly consistent with Lipinski compliance [17]. The prevalence of pyrimidine-containing fragments as optimal C-2 substituents for EGFR binding is a recurring theme in the broader medicinal chemistry literature on heterocyclic kinase inhibitors and is directly reflected in the structure of gefitinib itself, which contains an anilinoquinazoline (essentially a bicyclic pyrimidine analogue) core [3].
The uniform bioavailability score of 0.55 and the absence of PAINS and Brenk alerts across the series compare favourably with other reported thiazolidinone series, where PAINS alerts have occasionally been identified for compounds bearing reactive electrophilic moieties or catechol substructures. The toxicity class 4 designation for the majority of the test compounds is also consistent with published ProTox-II data for related scaffolds in the early discovery phase, and the absence of mutagenicity and cardiotoxicity predictions distinguishes this series from some related heterocyclic scaffolds that have shown genotoxic potential [14].
4.6 Limitations and Future Directions
This study has several limitations that should be acknowledged. The in-silico nature of the investigation means that all results are predictive rather than experimentally confirmed. Molecular docking scores are estimates of binding free energy that do not account for protein flexibility, solvation effects, or entropy contributions with full accuracy, and the correlation between docking scores and experimental IC50 values is rarely perfect [8]. The ADMET predictions from SwissADME and ProTox-II are based on computational models trained on existing drugs and chemical data, and their accuracy for novel scaffold classes may be limited by structural analogy gaps in the training datasets. In particular, ProTox-II hepatotoxicity and organ toxicity predictions carry model-specific uncertainties that necessitate experimental confirmation.
Future work should include the experimental synthesis of the designed compounds, particularly TD-4, followed by in vitro cytotoxicity evaluation using cancer cell lines such as A549 (lung adenocarcinoma) and MCF-7 (breast adenocarcinoma) using the MTT assay, EGFR kinase inhibition assays using recombinant enzyme, and cell-based EGFR phosphorylation studies. Molecular dynamics simulations over nanosecond timescales would provide more accurate estimates of binding stability and allow examination of induced-fit effects and binding mode stability that cannot be captured by rigid-receptor docking. The encouraging preliminary data from this in silico study, particularly for TD-4, provides a clear rationale for investing in these experimental validation studies.
CONCLUSION
This study has presented the rational design and comprehensive in silico evaluation of six novel 1,3-thiazolidin-4-one derivatives bearing a 3-(1,3-thiazol-2-yl) group as potential EGFR-targeted anticancer agents. Molecular docking against the EGFR kinase domain crystal structure (PDB ID: 1ZXM, 1.87 A resolution) demonstrated that all six compounds engage productively with the active site, with binding affinities ranging from -5.9 to -6.6 kcal/mol. Among the designed compounds, TD-4, bearing a pyrimidin-4-yl substituent at C-2, exhibited the best binding affinity (-6.6 kcal/mol) and the most extensive interaction network, forming hydrogen bonds and complementary contacts with seven active site residues including the catalytically critical ARG817, ASP813, and ASN818. These residues overlap significantly with those engaged by the reference standard gefitinib, providing a strong pharmacophoric basis for further optimisation.
All six thiazole derivatives fully complied with Lipinski's rule of five, showed a bioavailability score of 0.55 comparable to the standard drug, and were free of PAINS and Brenk structural alerts. ProTox-II predictions confirmed the absence of mutagenic, cytotoxic, and cardiotoxic potential for the entire series, with a generally manageable acute toxicity profile. The SAR analysis identifies the pyrimidine heterocycle at C-2 as the most productive substituent for EGFR engagement within this scaffold, while the aminophenyl and imidazolyl groups may require further structural refinement to improve both potency and safety.
Taken together, the in-silico data generated in this study establish TD-4 as the most promising lead compound within the thiazole series for anticancer drug development targeting EGFR. The results provide a rational and evidence-based foundation for the experimental synthesis and biological evaluation of TD-4 and related structural analogues, with the goal of identifying optimised thiazole-based EGFR inhibitors with improved potency relative to currently available clinical agents.
REFERENCES




Manoj Gangadhar Shinde, Sayali Valiba Bodake*, Omkar Sanjay Bodke, Pallvi Dhananjay Bodke, Computational Drug Design, Molecular Docking, Pharmacokinetic Profiling, And Toxicity Study Of Novel Thiazole Derivatives For Anticancer Activity Using In Silico Approaches, Int. J. Sci. R. Tech., 2026, 3 (6), 1058-1074. https://doi.org/10.5281/zenodo.20731706
 10.5281/zenodo.20731706
10.5281/zenodo.20731706