
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Rashtrasant Janardhan Swami College of Pharmacy, Kokamthan, Tal- Kopargaon, Dist. Ahilyanagar, Maharashtra, 423601, India
The formulation of poorly water-soluble drugs remains one of the most formidable challenges in the pharmaceutical industry, particularly with the increasing prevalence of BCS Class II and IV candidates in the drug discovery pipeline. Amorphous Solid Dispersions (ASDs) have emerged as a premier strategic platform to overcome these limitations by dispersing the drug in a molecularly disordered state within a polymer matrix. This review explores the critical role of drug–polymer synergy in governing the success of ASDs. We analyze the molecular mechanisms such as hydrogen bonding, ionic interactions, and hydrophobic effects that provide the necessary thermodynamic and kinetic stabilization to inhibit the natural tendency of amorphous drugs to undergo recrystallization. Furthermore, the paper highlights the strategic use of polymers in achieving polymorphic control, ensuring that the drug remains in its high-energy amorphous form throughout its shelf-life and during the critical phase of gastrointestinal supersaturation. By maintaining a "spring and parachute" effect, these synergistic interactions significantly enhance the apparent solubility and oral bioavailability of recalcitrant molecules. Finally, we discuss the impact of advanced manufacturing technologies and the selection criteria for specialized polymers in designing the next generation of stable, high-performance solid dispersions.
The pharmaceutical industry is currently facing a significant challenge: approximately 70% to 90% of new chemical entities (NCEs) emerging from drug discovery pipelines are classified as poorly water-soluble, falling into BCS Class II (high permeability, low solubility) or BCS Class IV (low permeability, low solubility) [1]. This low aqueous solubility leads to poor dissolution rates in the gastrointestinal tract, which ultimately results in inconsistent absorption and low oral bioavailability. Among the various solubilization strategies—such as lipid-based formulations, nanocrystals, and salt formation—Amorphous Solid Dispersions (ASDs) have emerged as one of the most successful and versatile platforms for enhancing the delivery of recalcitrant molecules [2].
An ASD is defined as a system where a poorly soluble drug is dispersed in a molecularly disordered state within a carrier matrix, typically a synthetic or semi-synthetic polymer. Unlike the crystalline state, where molecules are arranged in a highly ordered lattice with strong intermolecular forces, the amorphous state lacks long-range order. This disordered state possesses higher internal energy, enthalpy, and entropy, which translates to a "dissolution advantage." Upon contact with aqueous media, the amorphous drug does not need to overcome the high lattice energy of a crystal, allowing it to reach a state of supersaturation a concentration far exceeding its equilibrium solubility [3].
However, the amorphous state is inherently thermodynamically unstable. Over time, or upon exposure to moisture and heat, amorphous molecules tend to reorganize and revert to their more stable crystalline counterparts, a process known as recrystallization. This phase transformation is the "Achilles' heel" of ASDs, as it leads to the loss of the solubility advantage during storage or during transit through the gut [4].
This is where drug–polymer synergy becomes critical. The polymer is not merely a passive diluent; it acts as a strategic stabilizer. Through specific intermolecular interactions, polymers increase the Glass Transition Temperature (T g) of the system, thereby reducing molecular mobility (kinetic stabilization). More importantly, polymers can exert polymorphic control by selectively interacting with specific functional groups of the drug, hindering the nucleation and growth of crystalline lattices [5]. This review focuses on the molecular mechanisms governing this synergy and evaluates how these strategic platforms are engineered to ensure both physical stability and clinical efficacy.
Evolution and Classification of Solid Dispersions
The concept of solid dispersions was first introduced by Sekiguchi and Obi in 1961 as a method to reduce the particle size of poorly soluble drugs to a molecular level [6]. Over the decades, the technology has evolved through several generations. The first generation utilized crystalline carriers like urea or sugars, which often resulted in limited stability. The second generation introduced amorphous polymeric carriers (e.g., PVP, PEG), which significantly improved solubility but faced challenges with moisture-induced recrystallization.
The current state-of-the-art, or third generation, involves the use of specialized, "designer" polymers and surfactants that create a synergistic environment. These systems are engineered not only to dissolve the drug but to maintain a stable, "high-energy" disordered state through complex intermolecular networking [7]. This evolution reflects a shift from simple physical mixing to sophisticated molecular engineering, where the polymer is selected based on its ability to form a "glassy solution" with the specific drug candidate.
The Regulatory and Economic Drive
From a pharmaceutical development perspective, the drive toward Amorphous Solid Dispersions (ASDs) is fueled by both clinical necessity and economic factors. As the cost of bringing a New Chemical Entity (NCE) to market exceeds 2.6 billion USD, drug developers cannot afford to abandon potent molecules simply due to poor solubility [8]. ASDs provide a "rescue platform" for these molecules.
Furthermore, regulatory agencies like the FDA and EMA have increasingly approved ASDbased products (e.g., Kalydeco, Ivacaftor), proving that the stability challenges of the amorphous state can be managed through robust formulation science. The strategic use of polymers to provide polymorphic control ensures that the drug remains in its most bioavailable form throughout the product's shelf-life, satisfying the stringent stability requirements of the International Council for Harmonisation (ICH) guidelines [9].
Therapeutic Rationale: The "Spring and Parachute" Concept
The ultimate goal of drug–polymer synergy in ASDs is best described by the "Spring and Parachute" model. The amorphous drug provides the "Spring"—the initial, rapid surge in concentration to a supersaturated state upon dissolution. However, without stabilization, this concentration would quickly "crash" due to precipitation. The polymer acts as the
"Parachute," delaying recrystallization and maintaining high intraluminal concentrations for a sufficient duration to allow for systemic absorption [10]. Understanding the molecular forces that enable this "parachute" effect is central to designing effective therapeutic ASDs.
Thermodynamics and Physical Stability of the Amorphous State
The fundamental challenge in formulating ASDs lies in the high-energy nature of the amorphous state. Unlike the crystalline state, which sits at a global thermodynamic minimum, the amorphous form is metastable or unstable, possessing excess internal energy, enthalpy, and volume. This section explores the forces that drive recrystallization and the synergistic role of polymers in mitigating these risks.
The Thermodynamic Drive for Recrystallization
From a thermodynamic perspective, the transition from an amorphous state to a crystalline state is governed by the difference in Gibbs free energy (Delta G). At any temperature below the melting point (T_m), the crystalline form is more stable. The "solubility advantage" of the amorphous form is a direct result of this excess free energy; however, this same energy acts as a continuous driving force for the molecules to reorganize into a periodic lattice [11].
The Glass Transition Temperature (T_g) and Kinetic Stabilization
ASDs are typically "glassy" solids. The Glass Transition Temperature (T_g) is the temperature at which a hard, brittle "glassy" state transitions into a soft, rubbery state.
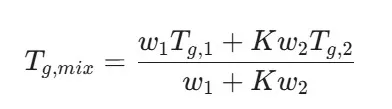
Where w represents the weight fraction and K is a constant related to the densities and thermal expansion coefficients of the components. Polymers, having high T_g values (e.g., PVP has a T_g of approximately 170°C), raise the T_{g,mix} of the ASD, ensuring it remains in the stable glassy state at room temperature [12].
Molecular Mobility and the "Hancock-Crowley" Prediction
The physical stability of an ASD is not just about the T_g, but the rate of molecular relaxation. Hancock and Crowley established that for an amorphous system to be considered "stable" for a shelf-life of two years, it should ideally be stored at a temperature at least 50°C below its T_g (T_{storage} < T_g - 50 K). This is known as the T_g - 50 rule [13].
Thermodynamic Miscibility and Solubility
A key aspect of drug–polymer synergy is miscibility. If a drug and polymer are not miscible, they will undergo "phase separation," creating drug-rich domains that recrystallize almost instantly. The Flory-Huggins Theory is used to describe the free energy of mixing (Delta G_{mix}):
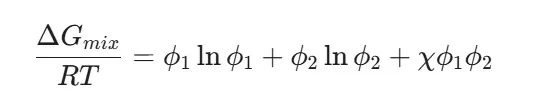
Where chi is the Flory-Huggins interaction parameter. A negative or low chi value indicates strong drug–polymer synergy, suggesting that the polymer can thermodynamically "dissolve" the drug molecules, preventing them from finding each other to form a crystal [14].
This brings us to the heart of your review paper. In Section 3, we move from the macrophysical properties (like T g ) to the micro-molecular level. To reach your 20-page goal, this section must be very descriptive regarding the chemical bonds that create "Synergy."
Molecular Mechanisms of Synergy: Intermolecular Interactions
The physical stability and polymorphic control of an ASD are not solely dependent on the "glassy" immobilization of the drug. Rather, they are significantly dictated by the formation of specific, high-affinity intermolecular interactions between the drug and the polymer. These interactions act as molecular "anchors," preventing the drug molecules from selfassociating into a crystalline lattice.
Hydrogen Bonding: The Primary Stabilizer
Hydrogen bonding is the most prevalent and influential interaction in ASD systems. It occurs when a hydrogen atom covalently bonded to an electronegative atom (like Oxygen or Nitrogen) in the drug molecule interacts with a lone pair of electrons on a functional group of the polymer (or vice versa).
Ionic Interactions and Salt Formation
In cases involving acidic or basic drugs, ionic interactions (coulombic forces) can provide even greater stabilization than hydrogen bonding.
pi-pi Stacking and Hydrophobic Interactions
For drugs that lack traditional hydrogen-bonding donors or acceptors (non-polar drugs), synergy is often achieved through pi-pi interactions or hydrophobic effects.
Spectroscopic Characterization of Synergy
To prove these interactions exist in your paper, you must discuss how they are measured. This adds professional depth:
Polymorphic Control and the Inhibition of Recrystallization
In the context of Amorphous Solid Dispersions (ASDs), the polymer serves as a strategic guardian against the thermodynamic "pull" toward the crystalline state. Recrystallization is typically a two-stage process: Nucleation (the birth of a crystal embryo) and Crystal Growth (the expansion of that embryo). Synergy between the drug and polymer can interrupt both stages, providing a robust mechanism for polymorphic control.
Inhibition of Nucleation
Nucleation occurs when drug molecules collide and align in a specific orientation to form a stable "nucleus." Polymers inhibit this through two primary mechanisms:
Retardation of Crystal Growth
Even if a nucleus forms, the polymer can prevent it from growing into a full-scale crystal.
Kinetic vs. Thermodynamic Stabilization
A critical distinction in your review is the difference between kinetic and thermodynamic stabilization.
Influence of Stereochemistry and Tacticity
To add advanced depth to your paper, we must discuss the "fit" of the polymer. The stereochemistry (arrangement of atoms) and tacticity of the polymer chains can influence how well they "interlock" with the drug molecule. A polymer with high stereoregularity may be less effective at maintaining the amorphous state than a random copolymer, which provides a more "disordered" environment that matches the drug's amorphous state [22].
Bioavailability Enhancement and the "Spring and Parachute" Effect
The ultimate objective of engineering drug–polymer synergy is to translate physical stability into enhanced therapeutic performance. For BCS Class II and IV drugs, the rate-limiting step for systemic absorption is aqueous solubility. ASDs bypass this by creating a state of supersaturation in the gastrointestinal (GI) fluids.
The Thermodynamic "Spring"
When an ASD is ingested, the high-energy amorphous drug is released into the GI tract. Because the drug lacks a crystalline lattice, it dissolves rapidly, reaching a concentration () that is much higher than its equilibrium crystalline solubility (). This rapid surge is referred to as the "Spring." This state creates a high concentration gradient across the intestinal membrane, which significantly increases the flux () of the drug according to Fick's First Law of Diffusion:
Where is the diffusion coefficient. A higher directly drives more drug into the bloodstream [23].
The Polymeric "Parachute"
Supersaturation is an unstable state; the drug naturally wants to precipitate or "crash" out of the solution to reach equilibrium. This is where the "Parachute" effect comes in.
Liquid-Liquid Phase Separation (LLPS)
In high-performance ASDs, the drug concentration may exceed a specific threshold known as the LLPS limit. At this point, the solution separates into a drug-poor aqueous phase and a
drug-rich colloidal phase (nanodroplets).
• Synergy Role: The polymer stabilizes these nanodroplets, preventing them from coalescing into large crystals. These nanodroplets act as a "reservoir," quickly replenishing the dissolved drug as it is absorbed through the intestinal wall [25].
Impact of Gastrointestinal pH and Transit
Strategic polymer selection allows for pH-targeted release. For instance, using enteric polymers like HPMCAS (Hypromellose Acetate Succinate) ensures the "Spring" is only triggered in the small intestine (pH > 5.5), preventing the drug from degrading or premature precipitation in the acidic environment of the stomach. This synergy between the drug's solubility and the polymer's pH-sensitivity is a cornerstone of modern ASD design [26].
Polymeric Carriers: Classification, Selection, and Manufacturing Impact
The success of an Amorphous Solid Dispersion (ASD) is fundamentally tied to the choice of the polymeric carrier. The polymer must not only be miscible with the drug but also compatible with the chosen manufacturing technology. This section evaluates the diverse landscape of pharmaceutical polymers and the high-throughput technologies used to produce stable ASDs.
Classification of Polymeric Carriers
Polymers used in ASDs are broadly classified based on their origin and their chemical functionality.
Synthetic Polymers
Semi-Synthetic (Cellulosic) Polymers
"Parachute" effect by maintaining supersaturation through steric stabilization.
Criteria for Polymer Selection
Selecting the right polymer is no longer a "trial and error" process. It involves a strategic evaluation of:
Manufacturing Technologies and Synergy Creation
The method of preparation is critical because it dictates how the drug and polymer are molecularly mixed.
Solvent-Based Methods (Spray Drying)
Spray drying involves dissolving the drug and polymer in a common solvent and rapidly atomizing the solution into a hot air stream. The "flash evaporation" of the solvent traps the drug in its disordered state within the polymer matrix before it has a chance to crystallize.
This method is preferred for thermolabile (heat-sensitive) drugs [29].
Fusion-Based Methods (Hot-Melt Extrusion - HME)
HME is a solvent-free, continuous process where the drug and polymer are melted and mixed under high shear. The intense mechanical energy forces the drug to "dissolve" into the molten polymer. HME is highly efficient for creating strong drug–polymer synergy, particularly for drugs that can form ionic bonds or strong hydrogen bonds with the carrier during the melting phase [30].
Future Perspectives
Emerging Trends in ASD Research
As the field of Amorphous Solid Dispersions (ASDs) matures, research is shifting from empirical "trial-and-error" methods to predictive, data-driven approaches. To maintain a competitive edge in pharmaceutical development, several emerging areas are gaining traction:
Addressing the Scale-Up and Stability Challenges
While the lab-scale success of ASDs is well-documented, the industry still faces hurdles in scale-up and global distribution. The sensitivity of amorphous systems to humidity (hygroscopicity) necessitates specialized packaging and climate-controlled supply chains, particularly for markets in tropical regions. Future research must focus on "moistureinsensitive" polymers and advanced coating technologies that provide a robust barrier without compromising the "Spring and Parachute" dissolution profile [34].
CONCLUSION
The development of Amorphous Solid Dispersions represents a paradigm shift in the management of poorly water-soluble drugs. By leveraging drug–polymer synergy, researchers can strategically overcome the thermodynamic instability of the amorphous state, providing a robust platform for polymorphic control and bioavailability enhancement.
The synergistic interplay—ranging from molecular-level hydrogen bonding to macro-level kinetic stabilization via T g elevation—ensures that the drug remains in its most bioavailable form. As we move toward the next generation of therapeutics, the convergence of advanced manufacturing like Hot-Melt Extrusion and predictive computational tools will solidify ASDs as a gold-standard platform for delivering the high-potency, low-solubility molecules of the future.
REFERENCES

Sakshi Ganesh Lokhande*, Sachin B. Aglave, Drug–Polymer Synergy In Amorphous Solid Dispersions: A Strategic Platform For Polymorphic Control And Bioavailability Enhancement, Int. J. Sci. R. Tech., 2026, 3 (7), 378-386. https://doi.org/10.5281/zenodo.21375843
 10.5281/zenodo.21375843
10.5281/zenodo.21375843