1Department of Molecular Biology, University of Okara, Punjab, 53600, Pakistan.
2Department of Biochemistry, Faculty of Life Science, University of Okara, Punjab, 53600, Pakistan
Conjunctival inflammation affects millions globally and can be triggered by microbial infections, allergens, or genetic factors. This study focuses on specific SNPs in the IL-4 gene, linked to pink eye susceptibility. Using In-Silico tools, 15 SNPs were identified as potentially damaging to IL-4 protein, with the R139G, A118G, R112C, and R139W variants showing probable damage on high RMSD values, as highlighted by SAVESV6.1. Structural analysis using HOPE predicted that the R139W mutation may be structurally bigger than the IL-4 wild type, while other variants were damaging to protein stability and function. Molecular docking studies with PyRx screened 25 small molecules for interactions with IL-4. Compounds such as Ioniflavone, Meridine, Palbociclib, Pyranoamentoflavone, Ramipril, and Tetracycline demonstrated strong binding affinities with IL-4, suggesting potential therapeutic efficacy. Visualization through Discovery Studio revealed differences in residue interactions, hydrogen bonds, and hydrophilic interactions between the wild-type and mutant forms, indicating that these SNPs could alter IL-4 structure and binding. These findings suggest that genetic variants in IL-4 may play a role in pink eye susceptibility and open avenues for developing targeted therapies. Their potential in targeting IL-4-related pathways further supports their use in addressing the immune component of conjunctivitis. However, each compound would require further clinical and pharmacological evaluation to confirm efficacy and safety for ophthalmic use. Further laboratory and clinical studies are essential to validate these results and explore personalized treatments for pink eye based on IL-4 genetic profiles.
Interleukin-4 (IL-4) is a type I pleiotropic cytokine known for its four-?-helical bundle structure and its wide-ranging effects on different cell types, which�is significant in both the innate and adaptive immune responses. Initially, B and T cells were identified as the primary targets affected by IL-4, highlighting its significance in immune regulation. In 1982, IL-4 was first discovered by Ellen Vitetta and her research team, with additional contributions by Maureen Howard and William E. Paul, who also helped characterize its functions [1]. �In humoral immunity, it directs B-cells to produce IgE while inhibiting Th1 cytokines (IFN-gamma and IL-2). This change highlights the significance IL-4 is for fostering a Th2-mediated, antibody-focused response, which is required for controlling extracellular infections and allergy responses [2]. The IL-4 receptor is widely distributed across various organs, including the brain, fibroblasts, endothelial cells, epithelial cells, muscles, and liver, where it regulates immune functions. Genetic mutations in IL-4 and IL-4R genes can alter protein expression and activity, potentially influencing susceptibility or resistance to infectious diseases, rheumatoid arthritis, asthma, and atopy [3]. �Elevated IL-4 levels are observed in seasonal allergic conjunctivitis patients and vernal keratoconjunctivitis, underscoring its significant role in these allergic eye conditions [4]. Conjunctival inflammation is frequently brought on by infections, allergies, or irritants, resulting in pink eye (conjunctivitis), which is characterized by redness in the eyes. Ocular discomfort, impaired vision, and light sensitivity are among the symptoms. Adenoviruses are frequently responsible for viral conjunctivitis [5], and are accompanied by fever, lymphadenopathy (especially preauricular), pharyngitis, and upper respiratory tract infections [6].
Moreover, common microbes responsible for bacterial conjunctivitis include Staphylococcus pneumoniae, Haemophilus influenzae, and Moraxella catarrhalis. Among these, H. influenzae, particularly non-typeable strains accounting for about 70% of cases [7] . Common signs of bacterial conjunctivitis include red eyes and a thick, mucopurulent discharge, often green or yellow [8]. Allergic conjunctivitis is a hypersensitivity reaction triggered by airborne allergens like pollen, dander, dust, and mold. It occurs when mast cells degranulate in response to an IgE-mediated reaction, releasing histamine and other inflammatory mediators. This condition peaks in young people between the ages of 15 and 18, affecting about 20% of them [9]. Similar to viral conjunctivitis, allergic conjunctivitis may show a follicular appearance of the tarsal conjunctiva, characterized by small bumps or nodules on the inner eyelid surface, along with watery discharge during clinical examination [10] Allergic conjunctivitis may not always show the typical symptoms, while blepharitis and meibomian gland dysfunction can present eye irritation and discomfort [11]. The IL-4 promotes the differentiation of naive T cells into Th2 cells, which boosts the production of immunoglobulin E (IgE) and other cytokines that drive allergic reactions and inflammation in the conjunctiva [12].
The IL-4 gene is located on human chromosome 5q31-q33 region and comprises 4 exons and 3 introns, with the exons encoding its functional domains. It is primarily produced by activated T cells, mast cells, basophils, and eosinophils that promote Th2 cell differentiation and stimulate B cell proliferation and class switching to IgE, playing a key role in humoral immunity and allergic responses. Immune responses and vulnerability to infections can be impacted by variations in the IL-4 gene [13]. The c.-589C>T (rs2243250) is located in the IL4 promoter region and shows notable differences in frequency between patients with viral conjunctivitis and healthy individuals [14]. The presence of the T allele may increase susceptibility to viral infections by altering IL-4 expression, potentially compromising immune responses [3]. The IL-4R variant rs1805010 (c.223A>G) was also analyzed for viral conjunctivitis, but no significant differences in its frequency were observed between affected patients and healthy controls [14].
It regulates B cell differentiation into plasma cells and promotes the proliferation of T and B cells. It induces B cells to switch to IgE production and enhances MHC class II expression. The IL-4 exposure also reduces the IL12 production, IFN?, and macrophages by dendritic cells and elevated IL-4 levels can lead to the development of allergic reactions [15]. In extravascular tissues, IL-4 promotes the activation of macrophages into M2 cells, which aid in tissue repair and immune regulation, while inhibiting M1 cells, which are involved in inflammation and host defense. Pathological inflammation decreases as M2 macrophages and the release of IL-10 and TGF-? increase, activated M2 cells produce substances (arginase, proline, polyamines, and TGF-?) that�are major contributors to wound healing and fibrosis [16]. Furthermore, IL-4 variants significantly impact human health by influencing immune responses related to eye infections, particularly viral conjunctivitis. This study explored the IL-4 protein using several bioinformatics approaches to predict pathogenic coding variants and evaluate their potential effects on disease. We performed a functional analysis to assess the variant's impact on protein stability and function. The 3D structure of IL-4 was predicted, and we analyzed the conformational changes caused by mutations.� Additionally, blind docking studies were conducted to investigate Protein-Ligand interactions, and the results were visualized to assess binding affinities and potential mechanisms. This comprehensive approach enhances our understanding of IL-4 role in pink eye disease.
� � � �
� � � � � � 
� � � �
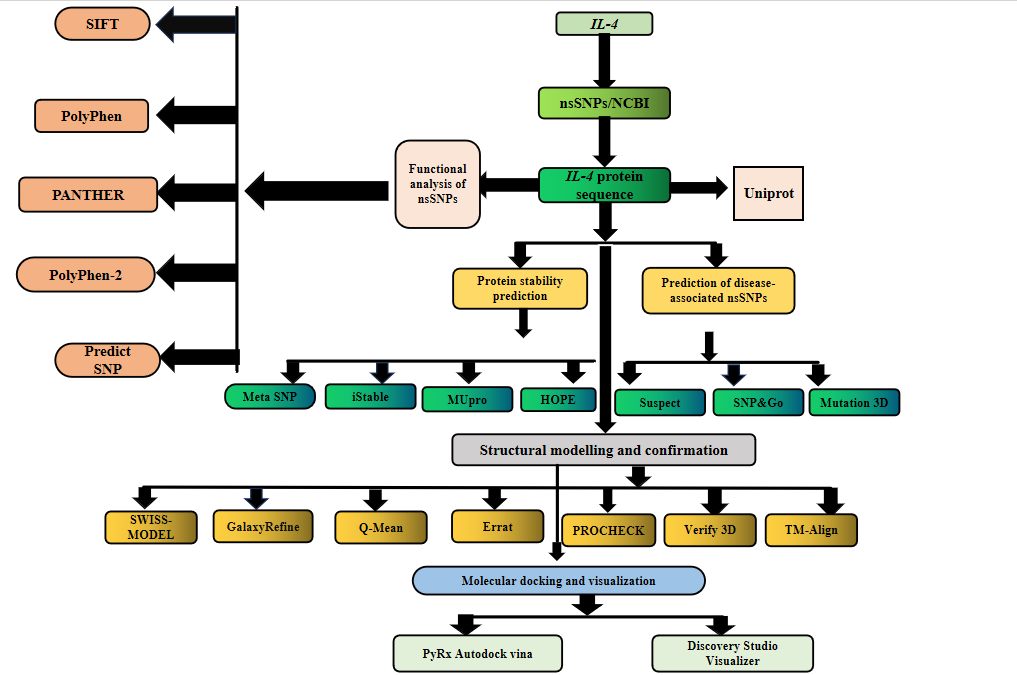
Figure 1. Schematic study of the�complete overflow of the current study
2.1. Variant collection
We collected data on the human IL-4 gene from the�National Center for Biotechnology Information (NCBI) (https://www.ncbi), GnomAD (https://gnomad.broadinstitute.org), Ensembl (http://asia.ensembl.org/Homo_sapiens/Gene/Summary), and the NCBI dbSNP database, which provided information on IL-4 gene variants. UniProtKB (http://www.uniprot.org/uniprot) was used to acquire the protein sequence in FASTA format. A schematic overview of the study workflow is provided in Figure 1.
2.2. Functional analysis of coding IL-4 variants
2.2.1. SNPnexus
We employed SNPnexus (https://www.snp-nexus.org) for functional predictions of IL-4 gene variants. The tool utilized rsIDs extracted from the dbSNP database as query inputs to assess the potential functional impacts of specific SNPs [17]. Sorting Intolerant from Tolerant (SIFT) tool based on sequence homology, evaluates mutation impacts on protein function. Mutations with a SIFT score of <�0.05 are predicted to be detrimental, while those with a score >0.05 are considered tolerable [18]. Polymorphism Phenotyping (PolyPhen) evaluates these variants based on structural data or a combination of sequence and structural information to predict their impact on protein function [18].
2.2.2. Polymorphism Phenotyping V2
PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2) played a crucial role in our investigation, enabling us to predict the potential implications of amino acid changes on protein structure and function. We entered our protein sequence, database ID/accession number, and details on amino acid changes into the server. Additionally, it provides a count score, with a value of 1 indicating the probable damage, along with predictions about the potential impact of missense variants on protein function [19].
2.2.3. Protein Analysis through Evolutionary relationship
PANTHER (http://pantherdb.org/tools/csnpScoreForm.jsp) predicts the functional impact of missense variants based on molecular activities, protein interactions, and evolutionary relationships. It calculates position-specific evolutionary conservation (PSEC) scores by aligning related proteins in structure and evolution. The prediction is based on the protein sequence and amino acid changes. The results are classified into three categories, probably damaging, possibly damaging, or probably benign based on evolutionary time and protein alignment data [20].
2.2.4. PredictSNP
PredictSNP (https://loschmidt.chemi.muni.cz/predictsnp1) combines data from multiple sources to predict the impact of a single amino acid change. By integrating various tools, it provides a more efficient and accurate consensus prediction compared to individual prediction methods [21].
2.3. Association of disease-causing SNPs
�2.3.1. Single nucleotide polymorphism & Gene Ontology
SNPs & GO (https://snps.biofold.org/snps-and-go/snps-and-go.html) uses a novel framework to predict whether a mutation is associated with disease, integrating genetic data, protein sequence, and Gene Ontology (GO) terms, along with 3D structural information. The tool classifies mutations as disease-associated if the probability score >0.5, and neutral <�0.5 while input files include FASTA sequences and protein variants [22].
2.3.2. Meta-SNP
Meta SNP (https://snps.biofold.org/meta-snp) is a meta-predictor that uses a random forest method to classify mutations as either 'Disease' or 'Neutral.' It is designed to detect disease-causing single nucleotide variations (nsSNVs), offering the�potential for early detection and classification of harmful genetic variations, which is a promising development in genomics [23].
2.3.3. SuSPect
SuSPect (http://www.sbg.bio.ic.ac.uk/suspect) combines annotation and sequence-based approaches to prioritize disease-candidate genes. By leveraging multiple lines of evidence, it enhances performance in scoring genes quickly and efficiently, while minimizing the impact of annotation bias, making it a valuable tool in genomic research for identifying disease-related genes [24].
2.4. Consequences of nsSNP on Protein Stability
2.4.1. iStable
The iStable server (http://predictor.nchu.edu.tw/iStable) predicts changes in protein stability by using either sequence or structure information as input. It integrates results from five different prediction tools, combining various methods developed by different teams to offer a more comprehensive and accurate assessment of potential changes in protein stability [25].
2.4.2. MUpro
The MUpro server (http://mupro.proteomics.ics.uci.edu) predicts the impact of single-site mutations on protein stability using machine learning tools, including neural networks and support vector machines. MUpro predictions provide the energy change (DDG), the direction of that change, and a confidence score (ranging from -1 to 1) to indicate the accuracy of the stability prediction [26].
2.5. Structural Integration of IL4
2.5.1. IL4 template prediction
The SWISS-MODEL server (https://swissmodel.expasy.org) predicts protein 3D structures using sequence data. By submitting a FASTA sequence, the server aligns it with templates from the UniProtKB proteome to generate a model. The quality of the resulting structure is assessed using various metrics, such as the Ramachandran Plot, the QMEAN score and the MolProbity score. Moreover, the server provides annotations with experimental data, enabling researchers to select the most reliable model for further analysis [27].
2.5.2. Model Validation and refinement
The QMEAN method (https://swissmodel.expasy.org/qmean/help) tool was used for qualitative model energy analysis. It evaluates the quality of protein models by comparing their z-scores to reference structures of similar size in the Protein Data Bank (PDB). This clustering-based approach provides an empirical solution, helping to assess the reliability of models by structural properties and their alignment with known reference data [28]. The 3D protein models were further assessed using the Structure Validation Server v6.1 (https://saves.mbi.ucla.edu), which provided an evaluation of the model's z-score and identified potential structural defects. This server offers a detailed analysis, helping to confirm the accuracy and stability of the selected model by highlighting any issues that could impact its reliability [29].
2.5.3. Template modeling alignment
The TM-align (https://zhanggroup.org/TM-align) is used to compare native and mutant protein structures by performing a residual-residual alignment. The comparison is quantified using the TM-score, which ranges between 0 and 1, and the RMSD (Root Mean Square Deviation), which measures the variation between the native and mutated structures. The TM-score indicates the structural similarity (1 represents perfect match), while the RMSD reflects the degree of variation or deviation between the two structures [30]. PyMOL (https://pymol.org) is assessed for visualizing, analyzing, and manipulating predicted residues and evolutionary sites in protein structure [31].
The gene sequences obtained from UniProtKB for the proteins are consistent, ensuring a reliable source for further bioinformatics analysis. The PubChem IDs of the pharmaceutical compounds used in this study were sourced from the ZINC database (https://zinc.docking.org) and PubChem (http://www.ncbi.nlm.nih.gov/pccompound). These compounds were then converted into PDB format using the Discovery Studio v4.1 (https://discover.3ds.com/discovery-studio-visualizer-download) and the academic edition of PyMol (https://pymol.org) [32, 33].
The docking experiments were carried out using the freely accessible PyRx 0.8 program (https://pyrx.sourceforge.io). The process began by loading a protein in PDB format and converting it into a macromolecule. A ligand was selected and transformed using the AutoDock button. The Vina Wizard was launched by clicking the start button, followed by setting up the grid with the "maximize" option. Vina was activated by pressing the appropriate button. After sufficient time, the best docking scores for each protein-ligand pair were recorded. To analyze the ligand-protein interactions, the Discovery Studio Visualizer was used to generate 2D and 3D PNG images of the ligand and protein [34, 35].
The Mutation 3D tool (http://www.mutation3d.org) was utilized to visualize and investigate the spatial distribution of amino acid alterations on protein structures and model [36]. The HOPE Project (http://www.cmbi.ru.nl/hope/input) is a comprehensive tool designed to forecast the physical and chemical properties of proteins, and their function, hydrophobicity, and spatial structure. Users can input a single identification, such as UniProt ID, Gene/Protein Symbol, or Ensemble Transcript, into its interface [37].
3.1. Datasets
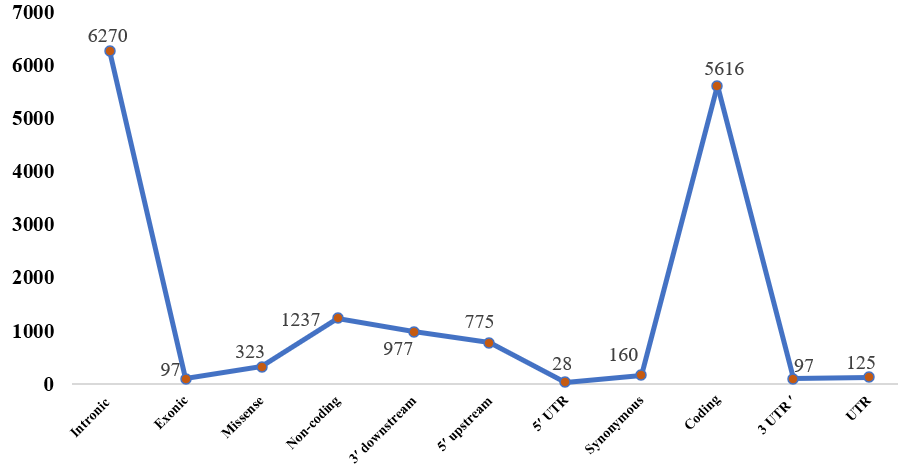
The variations in the human IL-4 gene were sourced from NCBI dbSNP, Ensembl, and GnomAD databases. Figure 2 categorizes single nucleotide polymorphism (SNP) distribution within the IL-4 gene, highlighting key genomic regions. The 3? UTR region contains 97 SNPs, and the broader UTR region has 125 SNPs. Within two distinct intronic regions, 5,133 SNPs are found in one, and 1,140 in the other. The coding region includes 5,616 SNPs, divided into 97 exonic, 323 missense, and 160 synonymous SNPs. Additionally, there are 1,237 SNPs in non-coding regions. The 3? downstream region contains 977 SNPs, while the 5? upstream region has 775 SNPs, including 28 SNPs within the 5? UTR. This detailed classification offers a comprehensive view of SNP distribution across the IL-4 gene. The missense nsSNPs were chosen for further investigation, as these variants are more likely to impact protein function by causing amino acid changes, potentially altering the structure, stability, or function of the IL-4 protein. This selection focuses on identifying variations with a higher probability of functional relevance, which may provide insights into the gene's role in disease mechanisms.
� � � �
� � � � � � 
� � � �
Figure 2. The prediction of variations is categorized into key genomic regions of IL4.
� � � �
� � � � � � 
� � � �
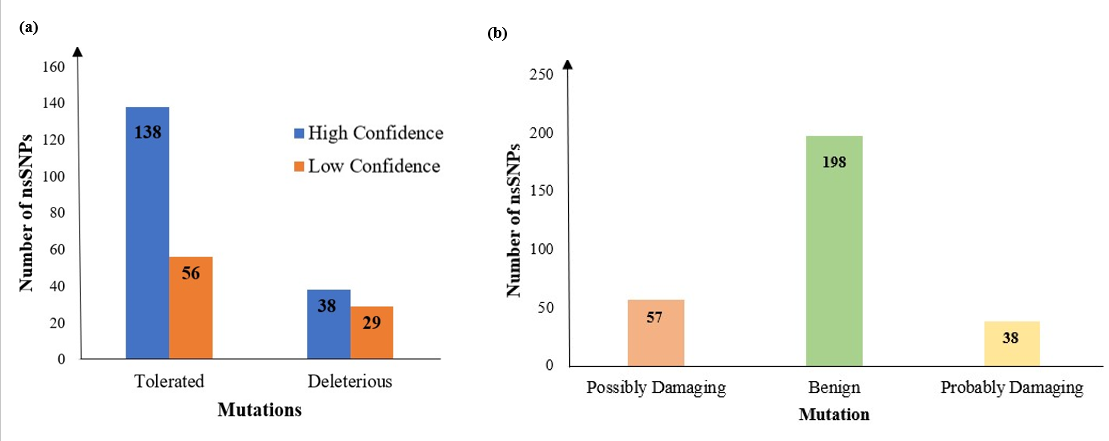
Figure 3. Bar plot of SNPs identified by a) SIFT and b) PolyPhen.
3.2. Deleterious nsSNPs In-Silico prediction
Using sequence homology of amino acids, SIFT provided predictions for a set of identified nsSNPs by revealing 261 nsSNPs as frequent, with 194 deletions projected to be deleterious and 67 deemed tolerable. PolyPhen results predicted that 57 nsSNPs were likely damaging, 38 possibly damaging, and 198 benign, as shown in Figure 3. Among these, 15 SNPs were identified as highly deleterious and selected for in-depth analysis as given in Table 1. For these 15 nsSNPs, predictions on functional impact and stability were performed using 10 different bioinformatic servers
Table 1. Prediction of 15 highly deleterious nsSNP found by SIFT and PolyPhen
|
Variation ID |
Variant |
Mutation |
SIFT |
Polyphen |
||
|
Score |
Prediction |
Score |
Prediction |
|||
|
rs1163858187 |
G/T |
C70F |
0 |
Deleterious |
1 |
Probably Damaging |
|
rs1446449750 |
C/A |
L76I |
0.01 |
Deleterious |
1 |
Probably Damaging |
|
rs149950065 |
C/A |
A118E |
0.01 |
Deleterious |
1 |
Probably Damaging |
|
rs778014138 |
G/A |
R139W |
0.01 |
Deleterious |
0.996 |
Probably Damaging |
|
rs199929962 |
T/C |
M144T |
0.03 |
Deleterious |
0.994 |
Probably Damaging |
|
rs1361163509 |
C/T |
A94T |
0.04 |
Deleterious |
0.994 |
Probably Damaging |
|
rs778014138 |
G/C |
R139G |
0.01 |
Deleterious |
0.993 |
Probably Damaging |
|
rs376367511 |
T/C |
C123R |
0 |
Deleterious |
0.99 |
Probably Damaging |
|
rs1444583505 |
T/C |
L7P |
0.02 |
Deleterious |
0.983 |
Probably Damaging |
|
rs149950065 |
C/G |
A118G |
0.02 |
Deleterious |
0.979 |
Probably Damaging |
|
rs775753738 |
C/T |
L110F |
0.01 |
Deleterious |
0.975 |
Probably Damaging |
|
rs753127489 |
C/A |
S81I |
0 |
Deleterious |
0.95 |
Probably Damaging |
|
rs751850550 |
G/A |
R112C |
0 |
Deleterious |
0.941 |
Probably Damaging |
|
rs1245723965 |
C/T |
A58V |
0.01 |
Deleterious |
0.934 |
Probably Damaging |
|
rs4252548 |
C/T |
R112H |
0.01 |
Deleterious |
0.922 |
Probably Damaging |
Furthermore, PolyPhen-2, PANTHER, and PredictSNP offer insights into the potential impact of mutations on IL4 protein function. Among the 15 nsSNPs analyzed, PANTHER predicted that 13 of these variants could be possibly damaging, while two (A94T and S81I) were classified as probably benign. PredictSNP results similarly identified A94T and S81I as neutral, with the remaining 13 nsSNPs likely to have deleterious effects. Moreover, PolyPhen-2 considered all 15 nsSNPs as probably damaging, as detailed in Table 2.
Table 2. Screening of�functional impact of nsSNPs by PANTHER, PredictSNP & Polyphen2
|
Variation ID |
Mutation |
PANTHER |
Predict SNP |
Polyphen2 |
|
|
Prediction |
Score |
Prediction |
Prediction |
||
|
rs1163858187 |
C70F |
Possibly damaging |
0.5 |
Deleterious |
Probably Damaging |
|
rs1446449750 |
L76I |
Possibly damaging |
0.5 |
Deleterious |
Probably Damaging |
|
rs149950065 |
A118E |
Possibly damaging |
0.5 |
Deleterious |
Probably Damaging |
|
rs778014138 |
R139W |
Possibly damaging |
0.5 |
Deleterious |
Probably Damaging |
|
rs199929962 |
M144T |
Possibly damaging |
0.5 |
Deleterious |
Probably Damaging |
|
rs1361163509 |
A94T |
Probably benign |
0.19 |
Neutral |
Probably Damaging |
|
rs778014138 |
R139G |
Possibly damaging |
0.5 |
Deleterious |
Probably Damaging |
|
rs376367511 |
C123R |
Possibly damaging |
0.5 |
Deleterious |
Probably Damaging |
|
rs1444583505 |
L7P |
Possibly damaging |
0.5 |
Deleterious |
Probably Damaging |
|
rs149950065 |
A118G |
Possibly damaging |
0.5 |
Deleterious |
Probably Damaging |
|
rs775753738 |
L110F |
Possibly damaging |
0.5 |
Deleterious |
Probably Damaging |
|
rs753127489 |
S81I |
Probably benign |
0.19 |
Neutral |
Probably Damaging |
|
rs751850550 |
R112C |
Possibly damaging |
0.5 |
Deleterious |
Probably Damaging |
|
rs1245723965 |
A58V |
Possibly damaging |
0.5 |
Deleterious |
Probably Damaging |
|
rs4252548 |
Hamna Tariq*, Aniqa Amir, Muhammad Saleem, Kainat Ramzan*, Tuba Aslam, Mehmooda Asif, In-Depth In-Silico Functional, And Structural Screening Of IL-4 Gene Variants Linked with Pink Eye Infection, Int. J. Sci. R. Tech., 2024, 1 (11), 213-229. https://doi.org/10.5281/zenodo.14234050 | ||||

 Hamna Tariq* 1
Hamna Tariq* 1
 10.5281/zenodo.14234050
10.5281/zenodo.14234050