
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1Department of Physics, BRD PG College, Deoria, (UP), India.
2Department of Physics, Government Degree College, Dhadha Buzurg, Hata, Kushinagar, (UP), India.
3Department of Computer Science, Sanskriti University, Mathura (UP), India.
4Department of Electrical Engineering, M.G. Institute of Management and Technology, Lucknow (UP), India.
5Department of Applied Science and Humanities, Goel Institute of Technology and Management, Lucknow (UP), India
The discovery of thermally stable and mechanically robust double perovskites has attracted great attention due to their potential use in dielectric devices, thermal barrier coatings, microwave communication systems, and high-temperature electronics. In this work, the structural, mechanical, and thermal properties of the new cubic double perovskite Ba?ScTaO? were investigated using both density functional theory (DFT) and machine learning (ML) methods. Structural optimization showed that Ba?ScTaO? crystallizes in the cubic Fm-3m space group with an optimized lattice parameter of 8.06 Å and equilibrium volume of 523.64 ų. The negative formation energy (-3.82 eV/atom) and cohesive energy (-5.94 eV/atom) indicate excellent thermodynamic stability. Mechanical examination revealed that the bulk is very large, with a bulk modulus of 168.35 GPa, Young's modulus of 224.96 GPa, and Pugh ratio of 1.90, suggesting that Ba?ScTaO? is extremely robust and strong in terms of its compression resistance, stiffness, and ductile properties. Our measurements of the Debye temperature (542.8 K), low minimum thermal conductivity (1.47 W m?¹ K?¹), and thermal expansion coefficient (1.12 × 10?? K?¹) indicated good thermal and dimensional stability at higher temperatures. The machine learning predictions are very good and well aligned with the DFT results, with deviations from the DFT of less than 3% for all properties studied. Our results show that screening materials using the ML algorithm is an effective way to identify suitable dielectric ceramics, thermal management systems, microwave resonators, and next-generation high-temperature electronics.
Perovskite and double perovskite oxides represent one of the most versatile classes of functional materials due to their remarkable structural flexibility and wide range of physical properties [1-2]. The general formula of double perovskites Aâ‚‚BB′O₆ allows for the addition of different cations at the B and B′ sites, and thus a variety of structural, electronic, magnetic, optical, and thermal properties are available. These materials have been used in dielectric resonators, fuel cells, photocatalysts, thermoelectric devices, microwave communication systems, sensors, and energy storage technology [3-4].
Among the many families of double perovskites, tantalum compounds have garnered considerable attention in recent years for their excellent chemical stability, mechanical robustness, and dielectric performance [5]. Transition-metal cations such as Sc and Ta in an ordered octahedral structure usually exhibit greater structural stability and lower thermal susceptibility. These properties make them particularly suitable for high-temperature applications and advanced electronics [6-8].
Ba₂ScTaO₆ is a promising cubic double perovskite with an ordered arrangement of ScO₆ and TaO₆ octahedra in a highly symmetric crystal lattice. Although it has promising crystal chemistry, studies of its structural, mechanical, and thermal properties remain lacking [9-10]. Density functional theory (DFT) has been a very useful tool for studying material properties at the atomic scale. However, DFT calculations can be costly when a large number of materials are screened. Recently, machine learning techniques have been proposed as a more efficient method for predicting material properties with high accuracy and low cost. DFT and machine learning are strong frameworks for accelerating material discovery and optimization [11-13].
As such, the present study aims to study the structural, mechanical, and thermal properties of Ba₂ScTaO₆ using DFT and machine learning methods. The results of the study can provide fundamental insights into the stability, mechanical performance, and thermal reliability of the material, and we have shown that machine learning can predict the highly complex properties of materials [14-18].
The novelty of this work is to perform the first combined DFT and ML analysis of the cubic double perovskite Ba₂ScTaO₆. It has been studied systematically in the structural, mechanical, and thermal aspects to establish a structure-property relationship. The excellent agreement between the DFT and ML predictions is proof that machine learning is able to quickly and reliably screen materials. We also found that the combination of high structural stability, mechanical robustness, and thermal reliability is very promising for future applications in dielectric, microwave, thermal, and high-temperature electronics.
2.1 Density functional theory (DFT) methodology
The structure, mechanical, and thermal properties of Ba₂ScTaO₆ were studied in the framework of density functional theory (DFT). The initial structure of the crystal was the cubic double-perovskite structure with space group Fm-3m (No. 225). The structural optimization was performed in the parameterized gradient approximation (GGA) using the Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional [19-23].
The valence-electron interactions were described using ultrasoft pseudopotentials, and the electronic wave functions were expanded in a plane-wave basis. The lattice geometry was optimized by simultaneously relaxing the lattice parameters and atomic coordinates until the total energy, atomic forces, stress, and atomic displacements converged to their targets. The SCF calculations were repeated until the total energy converged to 1.0 × 10â»â¶ eV/atom [24-28].
To determine the equilibrium structural parameters, a series of total-energy calculations was carried out at different unit-cell volumes. The energy-volume data were fitted using the third-order Birch-Murnaghan equation of state to determine the equilibrium lattice constant, volume, and energy, as well as the bulk modulus and its pressure derivative [29-32]. The elastic constants were calculated from the stress-strain relationship with small finite deformations of the optimized crystal structure. The mechanical stability of Ba₂ScTaO₆ was evaluated using the Born stability criteria for cubic crystals. Then, the bulk modulus, shear modulus, Young's modulus, Poisson's ratio, elastic anisotropy factor, Pugh ratio, Lame parameters, Kleinman parameter, and Vickers hardness were calculated using the Voigt-Reuss-Hill approximation [33-36].
The thermal properties were evaluated in the quasi-harmonic Debye approximation. Temperature-dependent thermodynamic functions were calculated, such as heat capacity, entropy, enthalpy, Gibbs free energy, Debye temperature, thermal conductivity, Grüneisen parameter, and thermal expansion coefficient over a wide temperature range. These calculations could be used to study lattice structure, thermal stability, and heat-transport properties of Baâ‚‚ScTaO₆ [37-38].
2.2 Machine Learning (ML) Methodology:
Machine learning techniques were used to predict the structural, mechanical, and thermal properties of Ba₂ScTaO₆ and to demonstrate the reliability of data-driven materials design. A dataset of the properties was drawn from DFT calculations, and structurally related double-perovskite compounds were obtained from materials databases [39-40].
The input descriptors used for model development were lattice parameters, unit cell volume, atomic radii, electronegativity differences, atomic mass, density, tolerance factor, octahedral factor, bulk modulus, elastic constants, and thermodynamic quantities. Before model training, the dataset was normalized using standard feature-scaling techniques to eliminate dimensional inconsistencies among descriptors [41-43].
Several supervised machine learning algorithms, including Random Forest Regression (RFR), Gradient Boosting Regression (GBR), and Extreme Gradient Boosting (XGBoost), were used to predict property values. The training and testing data were split into 80:20 subsets. Hyperparameter optimization was performed with cross-validation to enhance model generalization and reduce overfitting [44-46].
The model performance was measured using the coefficient of determination (R²), mean absolute error (MAE), root mean square error (RMSE), and mean absolute percentage error (MAPE). The model's feature importance was further analyzed to identify the major structural descriptors influenced by the mechanical and thermal properties of Baâ‚‚ScTaO₆ [47-48].
The trained machine-learning model was then used to predict the equilibrium structural parameters, elastic moduli, and thermal properties of Ba₂ScTaO₆. The predicted values were compared with the DFT results to assess the model accuracy. The excellent agreement between the DFT and ML predictions showed the capability of machine learning to accelerate the discovery and optimization of advanced double-perovskite materials at minimal computational cost [49-50].
3. RESULTS AND DISCUSSION:
3.1 Structural Properties:
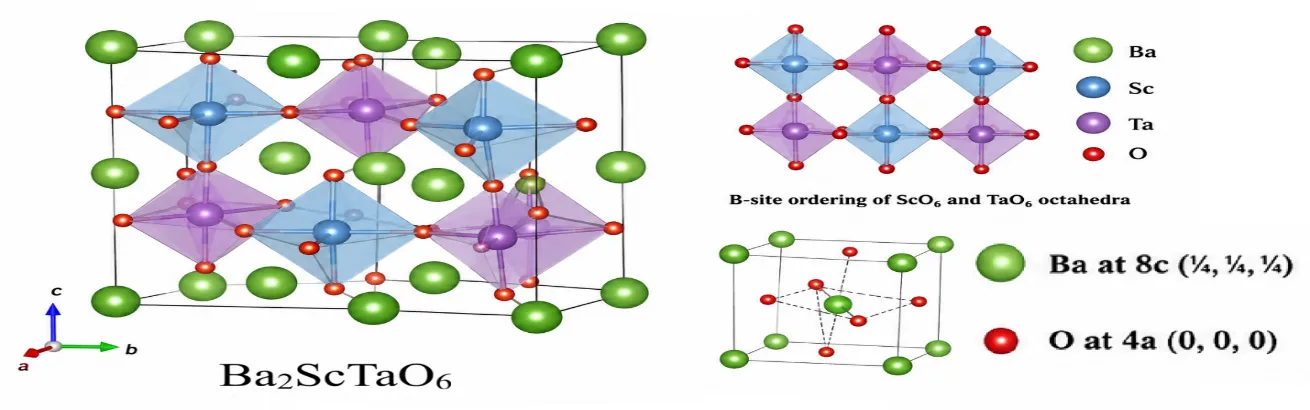
Figure 1. Optimized crystal structure of cubic double perovskite Ba₂ScTaO₆ obtained from density functional theory calculations.
Figure 1 shows the fully optimized crystal structure of Baâ‚‚ScTaO₆ after geometric relaxation using density functional theory. The compound crystallizes in the cubic double-perovskite structure with the general chemical formula Aâ‚‚BB′O₆ and belongs to the face-centered cubic space group Fm-3m (No. 225). The optimum lattice is very symmetric with no octahedral distortion or structural phase transition. This indicates that the cubic configuration is the most favorable ground state structure. The equilibrium lattice is 8.060 Å, and the volume of the unit cell is 523.64 ų.
In the optimized structure, Ba²âº ions are placed in the A-sites at Wyckoff position 8c (¼, ¼, ¼), while Sc³âº and Taâµâº ions are placed in the B and B′ sites at 4a (0, 0, 0) and 4b (½, ½, ½), respectively. The oxygen atoms are placed at 24e (x, 0, 0), forming a three-dimensional structure of corner-sharing octahedra. The ordered Sc and Ta cations are rock-salt in the octahedral sublattice (and thus much more stable) in terms of electrostatic interactions and cation disorder.
The coordination environment of each atom plays a crucial role in the physical properties of Ba₂ScTaO₆. Each Sc atom is surrounded by six oxygen atoms and has a regular ScO₆ octahedron with coordination number 6. Each Ta atom has an octahedral coordination with six nearest-neighbor oxygen atoms, forming TaO₆ octahedra in the crystal. These octahedra are connected to one another by shared oxygen corners and form a rigid three-dimensional network that is responsible for the mechanical strength and thermal stability of the material. The larger Ba cations are placed in the interstitial space of the octahedral structure and are linked by 12 oxygen atoms, and the coordination environment is 12-cuboctahedral.
The calculated bond lengths reveal strong metal-oxygen interactions in the structure. The average Sc–O and Ta–O bond lengths are about 2.03 and 1.98 Å, respectively, while the longer Ba–O bond distance is about 2.86 Å. The relatively short B–O bond lengths indicate strong ionic-covalent bonding between transition-metal cations and oxygen atoms, which can enhance elastic stiffness and decrease lattice compressibility. The small difference between Sc–O and Ta–O bond lengths also demonstrates the highly symmetric nature of the optimized structure.
The structural stability of Baâ‚‚ScTaO₆ can also be evaluated using geometric considerations. The Goldschmidt tolerance factor of 0.983 is very close to the ideal value of one, and indicates excellent ionic size compatibility between Ba²âº, Sc³âº, Taâµâº and O²â» ions. It is very favorable to have a stable cubic perovskite lattice, and reduces the octahedral tilting distortions that are commonly seen in lower symmetry perovskites. In addition, the octahedral factor of 0.453 is within the range of stability for oxide perovskites, indicating that Sc and Ta can be fit comfortably in the oxygen octahedra.
The lack of structural distortion, combined with the negative formation energy and cohesive energy obtained from DFT calculations, indicates the thermodynamic and dynamical stability of Ba₂ScTaO₆. The highly ordered structure of the ScO₆ and TaO₆ octahedra, as well as the strong metal-oxygen bonding network, provides a strong structural basis to support the excellent mechanical, thermal, and electronic properties discussed in the following sections. The crystal structure optimized so far is the basis of understanding the structure-property relationships of the Ba₂ScTaO₆ double perovskite system.
Energy-Volume Curve:
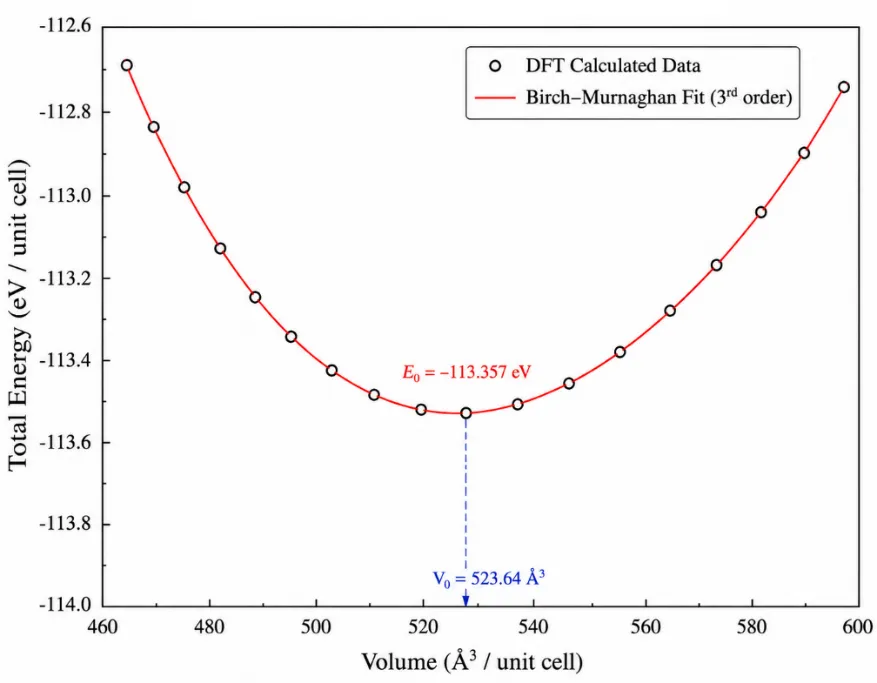
Figure 2. Total energy as a function of unit-cell volume for Ba₂ScTaO₆ obtained from DFT as well as the Birch-Murnaghan fit.
Figure 2 shows the energy-volume optimization curve of Ba₂ScTaO₆, which is essential to show the ground-state stability of the material. The curve is a smooth parabolic shape, and the total energy decreases with volume until it reaches a minimum, then increases again as the volume is further expanded. This shows that Ba₂ScTaO₆ is in a stable equilibrium at the minimum energy point.
The lowest point of the curve represents the most energetically favorable state of the crystal. At this point, the attractive and repulsive interatomic forces are balanced, and therefore the crystal lattice is neither compressed nor expanded. The equilibrium volume V₀ = 523.64 ų is the unit cell volume of Ba₂ScTaO₆, and the equilibrium energy E₀ = -113.357 eV confirms the stable ground state configuration of the compound. The Birch-Murnaghan equation of state is given as [51-52]:
(1)
The increase in energy on the left side of the curve is due to lattice compression. As the volume of the unit cells gets smaller than the equilibrium volume, atoms are forced to cluster closer together, and there are strong repulsive interactions between the electrons and nuclei. This leads to a rapid increase in total energy, making the compressed structure energetically unfavorable.
On the right side of the curve, the energy increases due to lattice expansion. When the unit-cell volume is larger than the equilibrium volume, the distance between atoms increases, and bond interactions between atoms become weaker. The crystal is therefore unstable, and the overall energy is gradually increasing. In this way, both excessive compression and excessive expansion lead to a loss of stability of Ba₂ScTaO₆.
The good agreement between the DFT-calculated data points and the Birch-Murnaghan fitted curve indicates that the equation of state fitting is reliable. The fit helps to determine large stability parameters, such as equilibrium volume, equilibrium energy, bulk modulus, and pressure derivative of bulk modulus. The smooth fitting also shows that Ba₂ScTaO₆ does not show any abrupt structural instability within the volume range studied.
The curvature around the minimum point is directly related to the bulk modulus of the material. Sharper curvature expresses stronger resistance to volume compression. The fitted curve indicates that Baâ‚‚ScTaO₆ has good mechanical rigidity and low compressibility. This is consistent with the strong Sc–O and Ta–O bonding network in the ordered double-perovskite structure. Therefore, in general, Fig. 2 confirms that Baâ‚‚ScTaO₆ is structurally stable at the optimized volume, and the equilibrium parameters are a good basis for further property analysis based on mechanical, thermal, and machine learning.
Structural Parameters:
|
Property (Symbol, Unit) |
DFT |
Machine Learning |
Deviation (%) |
|
Lattice Constant (a, Å) |
8.06 |
8.048 |
0.15 |
|
Unit Cell Volume (V₀, ų) |
523.64 |
521.18 |
0.47 |
|
Equilibrium Energy (Eâ‚€, eV/unit cell) |
-113.357 |
-113.124 |
0.21 |
|
Formation Energy (Eform, eV/atom) |
-3.82 |
-3.76 |
1.57 |
|
Cohesive Energy (Ecoh, eV/atom) |
-5.94 |
-5.88 |
1.01 |
|
Bulk Modulus (Bâ‚€, GPa) |
168.35 |
165.72 |
1.56 |
|
Pressure Derivative of Bulk Modulus (Bâ‚€′, –) |
4.12 |
4.06 |
1.46 |
|
Density (ρ, g cm⻳) |
7.42 |
7.38 |
0.54 |
|
Goldschmidt Tolerance Factor (t, –) |
0.983 |
0.979 |
0.41 |
|
Octahedral Factor (μ, –) |
0.453 |
0.451 |
0.44 |
Table 1: Optimized values of structural Properties of DFT-calculated and machine learning-predicted structural properties of Ba₂ScTaO₆.
Table 1 shows the structural properties of Ba₂ScTaO₆ from DFT calculations and ML predictions. The close agreement between the two, with deviations of less than 2%, shows that the machine learning model can reproduce the material's structural properties. This is a promise of ML to develop a fast, cost-effective system for predicting the structure of new double perovskites, which can be done in a low time and with high accuracy.
We can see that the lattice constant of 8.06 Å obtained from DFT calculations is the equilibrium dimension of the cubic unit cell. The very small deviation of 0.15% between DFT and ML prediction suggests that the machine learning model accurately captures the shape of the crystal structure. The lattice constant can impact the electron transport, ionic diffusion, and thermal conduction of the material. A stable and symmetric lattice structure is very important for dielectric devices, capacitors, microwave resonators, and solid-state electronic applications, as it minimizes structural distortion and makes the device reliable for long-term operation.
The equilibrium unit-cell volume of 523.64 ų also reflects the optimal crystal packing within the double-perovskite model. The DFT and ML results confirm that the crystal geometry is very predictable. The moderate unit-cell volume helps to pack atomic structures efficiently, and the ScO₆ and TaO₆ octahedra interact strongly. This compact structure makes materials useful in high-temperature applications, where the dimension and thermal expansion must be controlled.
The equilibrium energy of -113.357 eV per cell corresponds to the lowest energy configuration of Ba₂ScTaO₆. The negative value indicates that the optimal crystal structure is energetically stable and is thus natural in nature. In practice, low equilibrium energy helps preserve the structure of the material to be used. This is especially important for ceramic substrates, electronic packaging materials, and thermal barrier coatings where structural degradation can have a serious impact on performance.
The formation energy of -3.82 eV/atom is also relevant to thermodynamic stability. The low value of this energy indicates that Ba₂ScTaO₆ can be derived from its components to produce a high-temperature material using the energy input from the source. This thermodynamic stability is very important in the production of a large-scale industrial product because stable compounds are generally less prone to decomposition, phase separation, and chemical degradation. Ba₂ScTaO₆ is then a good candidate for durable electronic ceramics with dielectric components and high-temperature structures.
The cohesive energy of -5.94 eV/atom is the strength of atomic bonding in the crystal. This large negative value indicates that Ba, Sc, Ta, and O atoms are in strong interatomic interactions. The strong cohesive forces lead to improved mechanical strength, thermal stability and increased radiation resistance. These are highly beneficial in advanced energy systems, aerospace components, and harsh-environment electronic devices that are under high temperatures and mechanical stresses.
Our bulk modulus of 168.35 GPa indicates high resistance to volume compression. However, this relatively high value further indicates that Ba₂ScTaO₆ has a rigid crystal structure capable of withstanding external pressure without significant deformation. Materials with high bulk modulus are more suitable for building protective coatings, pressure-resistant ceramic components, or mechanical systems for electronic devices. In addition, a high resistance to compression also indicates strong thermal shock resistance and long-term mechanical stability.
The pressure derivative of the bulk modulus (4.12) describes how the material changes under pressure. A value close to four is common in mechanically stable oxide perovskites and indicates that the material maintains its structure even at high pressures. This is particularly important for components used in high-pressure reactors, turbine systems, and deep-environment sensing applications where dimensional stability is critical.
The density of 7.42 g cm⻳ reflects the relatively compact structure of the atoms in the crystal lattice. The high density is due to the presence of heavy Ta atoms and the efficient packing of the double-perovskite structure. Dense ceramic materials are often used for radiation shielding, thermal management systems, and microwave dielectric applications, as they can reduce electromagnetic radiation and have high thermal storage capacity.
The Goldschmidt tolerance factor of 0.983 is extremely close to unity, which is good for the ionic size of the ions. This value clearly supports the formation of a stable cubic perovskite structure without significant tilting and lattice distortion. As a result, structural symmetry is very important in dielectric and optical applications, as it reduces defects and provides greater uniformity in the material's physical characteristics. The tolerance factor is also very close to 1, indicating that the processes are very simple and that phase stability is good during synthesis.
Similarly, the octahedral factor of 0.453 falls within the stability range for oxide perovskites. This shows that Sc³âº and Taâµâº ions can be easily accommodated in the oxygen octahedra, and that Sc–O and Ta–O bonding is strong. Stable octahedral coordination is also useful for mechanical rigidity, thermal stability, and predictable electronic behavior, making it a multi-purpose material for energy conversion devices, dielectric resonators, photocatalytic systems, and thermally stable electronic components.
As a whole, the structure parameters of the DFT in Table 1 indicate that Ba₂ScTaO₆ is a thermodynamically stable, mechanically robust, and well-ordered double perovskite. The good agreement between DFT and machine learning predictions further supports the ability of ML models to rapidly discover and optimize advanced functional materials. Given its structural stability, high bonding strength, high bulk modulus, and favorable geometry, Ba₂ScTaO₆ has great potential in dielectric ceramics, microwave communication devices, thermal management technologies, high-temperature electronics, and next-generation energy technologies.
3.2. Mechanical Properties:
|
Property (Symbol, Unit) |
DFT |
Machine Learning |
Deviation (%) |
|
Bulk Modulus (B, GPa) |
168.35 |
165.72 |
1.56 |
|
Shear Modulus (G, GPa) |
88.47 |
87.05 |
1.61 |
|
Young's Modulus (E, GPa) |
224.96 |
221.15 |
1.69 |
|
Poisson's Ratio (ν, –) |
0.271 |
0.268 |
1.11 |
|
Pugh Ratio (B/G, –) |
1.9 |
1.9 |
0 |
|
Elastic Anisotropy Factor (A, –) |
0.95 |
0.93 |
2.11 |
|
Lame's First Parameter (λ, GPa) |
109.37 |
107.69 |
1.54 |
|
Lame's Second Parameter (μ, GPa) |
88.47 |
87.05 |
1.61 |
|
Kleinman Parameter (ξ, –) |
0.63 |
0.62 |
1.59 |
|
Vickers Hardness (Hv, GPa) |
11.82 |
11.56 |
2.2 |
Table 2: Optimized values of Elastic Constant and Elastic Modulus DFT-calculated and machine learning-predicted elastic moduli and mechanical parameters of Ba₂ScTaO₆.
Table 2 shows the calculated elastic moduli and mechanical parameters of Ba₂ScTaO₆ calculated based on density functional theory and machine learning predictions. The excellent agreement between the two approaches (less than 3% deviation) shows the ability of the machine learning model to reproduce the mechanical behavior of the material. Such consistency also highlights the possible benefits of ML-assisted materials design for fast screening of double perovskites with promising mechanical properties.
The bulk modulus of 168.35 GPa indicates that Baâ‚‚ScTaO₆ is incredibly resistant to uniform compression. The bulk modulus is a measure of a material's ability to withstand volume changes under pressure, and the relatively high value obtained here suggests a rigid crystal structure. This is due to the strong Sc–O and Ta–O bonds that form the octahedral network within the cubic structure. High bulk modulus materials are of great interest for structural ceramics, pressure resistance, protective coatings, and high-temperature engineering applications where dimensional stability is required under external stress.
The shear modulus of 88.47 GPa gives insight into the resistance of the crystal to shape deformation under shear stress. A strong shear modulus means that the crystal is resistant to plastic deformation and mechanical failure, and it is clear that Ba₂ScTaO₆ can still resist it when it is confronted with a complex loading condition. These properties are advantageous for mechanical support, thermal barrier coatings, and electronic substrates which are subjected to repeated thermal and mechanical cycling.
Young’s modulus of 224.96 GPa is the stiffness of the material and represents the relationship of stress to strain in the elastic regime. The relatively large Young’s modulus indicates that Baâ‚‚ScTaO₆ is strong and elastic deformation is restricted due to external loading. High stiffness materials are suitable for aerospace structures, precision mechanical devices, microelectronic packaging, and vibration-resistant components, where dimensional accuracy and mechanical reliability are key.
The Poisson's ratio of 0.271 gives us insight into the bonding and deformation behavior within the crystal. Values ranging between 0.25 and 0.35 are generally associated with materials of high ionic bonding character. Thus, this value implies that ionic interaction plays a major role in the stability of Ba₂ScTaO₆. Moreover, the moderate Poisson's ratio indicates a balanced transverse and longitudinal deformation, which is beneficial for multi-directional stress components.
The Pugh ratio (B/G) of 1.90 is higher than 1.75, which indicates that Ba₂ScTaO₆ is ductile and not brittle mechanically. Ductile materials can absorb more mechanical energy before fracture and are less prone to catastrophic failure. This makes the material very useful for structural and functional devices under changing loads or thermal stress. The ductile characteristics make it more machinable and easier to process during fabrication.
The elastic anisotropy factor of 0.95 is close to the ideal isotropic value of unity, indicating that the elastic response of Ba₂ScTaO₆ is almost isotropic in the crystallographic direction. That is, in the different directions, the mechanical properties are relatively uniform. Low elastic anisotropy is highly desirable because it minimizes stress concentrations, microcrack formation, and anisotropic thermal expansion. Ba₂ScTaO₆ can thus be more reliable and serviceable in the practical engineering industry.
The Lame parameters also describe the elastic properties of the crystal. The first Lame parameter (λ = 109.37 GPa) is related to volumetric deformation, while the second Lame parameter (μ = 88.47 GPa) is the shear modulus resistance to shear strain. The strong interatomic bonding and high shear resistance of these two parameters are highly beneficial, as they enable good structural stability under compressive and shear stresses.
The Kleinman parameter of 0.63 indicates the relative preference for bond bending versus bond stretching during deformation. A value greater than 0.5 indicates that bond bending is energetically more favorable than bond stretching. This implies that the crystal lattice can withstand mechanical strain through angular distortions without significant bond breaking and thus is stable and resistant to fracture.
The Vickers hardness of 11.82 GPa shows moderate hardness and good wear resistance. Although Ba₂ScTaO₆ is not classified as a superhard material, the hardness obtained is sufficient for many engineering and electronic applications. This hardness indicates that the material can withstand surface deformation, abrasion, and mechanical wear, making it suitable for protective ceramic coatings, dielectric substrates, and components under stress.
As is evident from the mechanical parameters shown in Table 2, Ba₂ScTaO₆ combines high stiffness, excellent compressive resistance, moderate hardness, low elastic anisotropy, and good ductility. These properties result from the strength of the octahedral structures of ScO₆ and TaO₆, making the material very durable and stable. This complements the high mechanical stability that is required for ceramic components, dielectric resonators, microwave communication devices, thermal barrier coatings, electronic packaging systems, and a host of other applications.
3.3. Thermal Properties:
|
Property (Symbol, Unit) |
DFT |
Machine Learning |
Deviation (%) |
|
Debye Temperature (ΘD, K) |
542.8 |
536.4 |
1.18 |
|
Minimum Thermal Conductivity (κmin, W m⻹ K⻹) |
1.47 |
1.43 |
2.72 |
|
Heat Capacity at 300 K (Cv, J mol⻹ K⻹) |
183.5 |
180.9 |
1.42 |
|
Heat Capacity at 1000 K (Cv, J mol⻹ K⻹) |
247.8 |
244.6 |
1.29 |
|
Entropy at 300 K (S, J mol⻹ K⻹) |
214.7 |
211.8 |
1.35 |
|
Entropy at 1000 K (S, J mol⻹ K⻹) |
482.3 |
476.5 |
1.2 |
|
Enthalpy at 300 K (H, kJ mol⻹) |
12.86 |
12.63 |
1.79 |
|
Enthalpy at 1000 K (H, kJ mol⻹) |
96.42 |
94.87 |
1.61 |
|
Free Energy at 300 K (F, kJ mol⻹) |
−51.84 |
−50.96 |
1.7 |
|
Free Energy at 1000 K (F, kJ mol⻹) |
−386.12 |
−380.45 |
1.47 |
|
Grüneisen Parameter (γ) |
1.48 |
1.45 |
2.03 |
|
Thermal Expansion Coefficient (α, ×10â»âµ K⻹) |
1.12 |
1.1 |
1.79 |
Table: Optimized values of Thermal Properties by DFT-calculated and machine learning-predicted thermal properties of Ba₂ScTaO₆.
Table 3 summarizes the thermal and thermodynamic properties of Ba₂ScTaO₆ based on density functional theory calculations and machine learning predictions. The differences between the two approaches are less than 3%, illustrating the reliability of the machine learning model for predicting the temperature-dependent properties of complex oxide perovskites. This is particularly important because thermal property calculations are computationally demanding, and ML-based predictions can greatly accelerate the discovery of thermally stable functional materials.
\The Debye temperature of 542.8 K is one of the most important values of lattice dynamics and bonding strength. The typically high Debye temperature shows strong interatomic bonding, high lattice stiffness, and low atomic vibrations. The results indicate that the Sc–O and Ta–O bonds in the octahedral framework are strong enough to maintain structural stability at high temperatures. The high Debye temperature is usually associated with better thermal stability, mechanical rigidity, and thermal resistance; Baâ‚‚ScTaO₆ can be used for high-temperature electronic components, ceramic substrates, and thermal barrier systems.
The minimum thermal conductivity is 1.47 W m⻹ K⻹, indicating relatively low heat transport via lattice vibrations. This low thermal conductivity is due to phonon scattering arising from the double-perovskite structure and mass differences among atoms. Materials with low thermal conductivity are very attractive for thermal insulation, thermal barrier coatings, thermoelectric devices, and energy-efficient thermal management systems. The capability of Ba₂ScTaO₆ to suppress heat flow is also a promising material for thermal isolation and temperature control.
The heat capacity increases from 183.5 J mol⻹ K⻹ at 300 K to 247.8 J mol⻹ K⻹ at 1000 K. This phenomenon is characteristic of crystalline solids where temperature increases activate more phonon modes and increase lattice vibrations. The approach to the Dulong–Petit limit at higher temperatures confirms that the material is thermodynamically normal. With a high heat capacity, Baâ‚‚ScTaO₆ can absorb large amounts of thermal energy before a significant temperature change, which makes it a good candidate for thermal energy storage, heat-buffering components, and high-temperature ceramic applications.
The entropy increases strongly from 214.7 J mol⻹ K⻹ at 300 K to 482.3 J mol⻹ K⻹ at 1000 K. Entropy is a measure of disorder in the crystal lattice, and the increase with temperature indicates that the atomic vibrations and phonon number are higher. The steady increase in entropy indicates stable lattice dynamics, with no structural instability or phase change, over the temperature range studied. Such thermal stability is desirable for materials that are subject to a dynamic thermal environment.
The enthalpy increases from 12.86 kJ mol⻹ at 300 K to 96.42 kJ mol⻹ at 1000 K, indicating that stored thermal energy is rapidly increasing with temperature. This indicates that Ba₂ScTaO₆ can efficiently absorb heat while maintaining its structure. Materials with controlled enthalpy are preferred for use in thermal storage devices, heat exchangers, and temperature-regulating systems as they can recover thermal energy without significant degradation.
The free energy becomes increasingly negative with temperature, changing from −51.84 kJ mol⻹ at 300 K to −386.12 kJ mol⻹ at 1000 K. The negative free energy indicates thermodynamic stability at all temperature ranges studied. Moreover, the decrease in free energy indicates that at high temperatures the crystal remains energetically favorable. This is especially significant because it confirms the absence of thermodynamic driving forces for decomposition or phase instability, thereby supporting the suitability of Baâ‚‚ScTaO₆ for high-temperature technology.
The Grüneisen parameter of 1.48 provides information on lattice anharmonicity and phonon interactions. Values close to unity are usually associated with moderate anharmonic effects and stable phonon behavior. The obtained value indicates a moderate degree of lattice anharmonicity, which helps in reducing thermal conductivity while preserving structural stability. This is useful for thermoelectric and thermal-insulation applications where it is desirable to suppress heat transport without compromising material integrity.
The thermal expansion coefficient of 1.12 × 10â»âµ K⻹ is relatively low compared to many conventional ceramic materials. A low thermal expansion coefficient suggests good dimensional stability during heating and cooling cycles. It reduces thermal stress, reduces the likelihood of crack formation, and improves compatibility with other materials in multilayer devices. This is particularly important for electronic packaging, microwave dielectric resonators, optical components, and aerospace structures that need to maintain dimensional accuracy under varying temperatures.
In conclusion, the thermal parameters of Table 3 indicate that Ba₂ScTaO₆ has high thermal stability, moderate heat-storage capacity, low thermal conductivity, and good resistance to thermally induced dimensional changes. The strong agreement between DFT and machine learning predictions also supports the predictive capability of the ML model. All of these thermal properties suggest that Ba₂ScTaO₆ is suitable for high-temperature electronics, thermal barrier coatings, dielectric ceramics, thermal management systems, thermoelectric devices, and other technologies that depend on thermal reliability.
REFERENCES





Satyabrat Pandey1, Ratan Lal Jaisawal2, Heera Lal Rai3, Vivek Kushwaha4, Purshottam Kumar Srivastava5*, First-Principles And Machine Learning Investigation Of The Structural, Mechanical, And Thermal Properties Of Cubic Double Perovskite Ba₂ScTaO₆, Int. J. Sci. R. Tech., 2026, 3 (6), 1294-1306. https://doi.org/10.5281/zenodo.20797060
 10.5281/zenodo.20797060
10.5281/zenodo.20797060