
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Kamla Institute of Pharmaceutical Sciences, Shri Shankaracharya Professional University, Bhilai, Chhattisgarh, India, 490020
Objective: In the present work, a series of new 1-(substituted phenyl)-N-(2-(2-(pyridin-2-yloxy) ethoxy) ethyl) methanimine has been designed, and a computational study has been performed on its possible efficacy in controlling postprandial hyperglycemia as an ?-glucosidase inhibitor. Materials and Methods: Twenty-four molecules were designed in ChemDraw and optimized in Chem3D in a series. ArgusLab 4.0 was used to perform molecular docking of the compound against ?-glucosidase (PDB ID: 3A4A) benchmarked with Miglitol. Discovery Studio Visualizer was used to visualize the interaction between the Ligands and proteins. The in silico pharmacokinetic properties, drug-likeness, and toxicity properties were calculated by SwissADME, Molsoft, and ProTox-II. Results and Discussion: All of the designed derivatives have higher binding affinities, ranging from -8.30 to -10.96 kcal/mol, than that of the parental drug Miglitol (with a binding affinity of -7.55 kcal/mol). These were M1 (-10.96 kcal/mol), M14 (-10.43 kcal/mol), M6 (-10.40 kcal/mol), and M22 (-10.20 kcal/mol) that exhibited the most favorable binding energies with stable hydrogen bonding and hydrophobic interactions with the key catalytic residues, ARG442, GLN279, THR306, and ASP69. The molecular weights, gastrointestinal absorption among derivatives, as well as drug-likeness scores for most, and the absence of high acute toxicity suggest that they possess good, predictable drug-like properties for oral intake. Conclusion: The computational results indicated that the effort designed methanimine derivatives are highly promising in terms of ?-glucosidase binding activity and have a good pharmacokinetic and safety profile. In summary, compounds M1, M6, M14, and M22 were identified as potential lead compounds and selected for further synthesis, biological evaluation, and optimization of their structures to develop new anti-diabetic drugs.
Diabetes mellitus (DM) is a chronic metabolic disorder characterized by persistent hyperglycemia resulting from impaired insulin secretion, defective insulin action, or a combination of both. Type 2 diabetes mellitus (T2DM), the more prevalent type of diabetes, has been linked with genes, obesity, a sedentary lifestyle, and unhealthy eating, but with Type 1 diabetes, the absolute insulin deficiency occurs from immune-mediated destruction of pancreatic β-cells. In addition, gestational diabetes mellitus is another health-related condition that can happen in pregnant women, thereby highlighting the importance of developing preventive and therapeutic measures [1,2]. In 2021, there were about 537 million people with diabetes globally; this is expected to rise to 643 million in 2030 and to 783 million in 2045, according to the International Diabetes Federation (IDF). Furthermore, there are almost 240 million people who have not had a diagnosis, which, apart from the fact of the delayed diagnosis, leads to possible worsening of the condition and more serious repercussions. In 2021, the global incidence of diabetes was estimated at approximately 966 billion USD spent across the healthcare system, which will further rise significantly in the next decade [3,4].
Chronic hyperglycemia leads to both microvascular and macrovascular complications, such as diabetic nephropathy, retinopathy, neuropathy, cardiovascular disease, stroke, peripheral vascular disease, and cognitive impairment, which markedly impact patients' quality of life and enhance mortality [5-7]. Controlling postprandial hyperglycemia has become an important therapeutic strategy for the management of T2DM. Significant attention has been directed at inhibiting digestive enzymes (DEs) that can break down dietary carbohydrates into absorbable glucose molecules; among them, α-amylase and α-glucosidase. α-Amylase breaks down starch into oligosaccharides, while α-glucosidase releases glucose from the terminal disaccharides and oligosaccharides within the brush border of the small intestine [8-10]. The ability to selectively inhibit α-glucosidase is effective in delaying glucose absorption, lowering the glucose level in the blood after a meal, and has proven to be an effective treatment strategy in diabetes. The clinically approved α-glucosidase inhibitors acarbose, miglitol, and voglibose have been proven to be effective in the long-term treatment of diabetes, but gastrointestinal upsets from abdominal discomfort, diarrhea, bloating, and flatulence were common with the long-term use of these medications. The restrictions have encouraged significant research effort in the development of new α-glucosidase inhibitors with greater efficacy, increased selectivity, better pharmacokinetic properties, and fewer side effects such as hypoglycemia [11].
Computational drug discovery methods have also significantly advanced the lead identification phase in recent years, allowing for the fast evaluation of molecular interactions, binding affinity, drug-likeness, and potential drug pharmacokinetics, before actual experimentation. Schiff bases (methanimine derivatives) are an important class of compounds containing N and have been reported to possess significant bioactivities of the following types: antimicrobial, anticancer, anti-inflammatory, antioxidant, and antidiabetic. The azomethine (-CH=N-) functionality provides for structural versatility and the ability to support favorable interactions with biological macromolecules. Similarly, scaffolds with a pyridine structure have been the subject of intense research in Medicinal Chemistry, due to their ability to form hydrogen bonds with the unfolded enzyme, to interact with enzyme active sites via π–π stacking and hydrophobic interactions, thus increasing the biological activity and the pharmacokinetic profile [12]. Further, replacing the aromatic ring can make enzymes more affined with the modification of structural properties, leading to the possibility of using substituted pyridine-based Schiff bases as promising candidates for the development of antidiabetic drugs. Molecular docking has become an efficient computer-aided drug design tool to predict the binding sites of molecules, their binding free energy, and the molecular recognition, which also helps in identifying the drug molecule candidates more rapidly. Combined with pharmacokinetic, drug-likeness, and ADMET analyses, it enables the early assessment of the therapeutic potential and oral bioavailability of newly designed compounds while reducing the time and cost of drug discovery. In this study, a series of novel 1-(substituted phenyl)-N-(2-(2-(pyridin-2-yloxy) ethoxy) ethyl) methanimine derivatives were designed, in silico pharmacokinetic properties were predicted, and molecular docking was done as a primary screen to assess the inhibitory activity towards α-glucosidase to develop them as potential inhibitors of α-glucosidase. This aim was to find molecules with good binding affinity that also possess good drug-like features to act as potential antidiabetic agents. The computational results are expected to supply a scientific foundation for the future synthesis of the most promising derivatives, their structural characterization, and biological evaluation.
2. MATERIALS AND METHODS
2.1 Software and Online Databases
An in-silico study utilized ChemDraw Ultra 22.2.0 and Chem3D 22.2.0 for the design of the ligands, ArgusLab 4.0.1 for molecular docking, SwissADME for pharmacokinetics assessment, and ProTox-II for checking the toxicity of the designed derivatives.
2.2 Molecular Docking Simulation
2.2.1 Ligand Preparation
The novel designed 1-(substituted phenyl)-N-(2-(2-(pyridin-2-yloxy) ethoxy) ethyl) methanimine (M1-M24) structures were sketched using ChemDraw software. The 2D structures were subsequently optimized to 3D structures, and the energy was minimized to find stable structures. The optimized structures of the ligands were stored in .pdb format for docking calculations [13, 14].
|
Compound ID |
R |
Compound ID |
R |
|
M1 |
-H |
M13 |
3-OCH3, 4-OH, 5-Br |
|
M2 |
4-ClD |
M14 |
4-CH3, 5-OH |
|
M3 |
3-Cl, 5NH2 |
M15 |
3-Br, 5-OH |
|
M4 |
4-F, 5-Cl |
M16 |
5-OH |
|
M5 |
3-Br, 5NO2 |
M17 |
4-NO2, 5-OCH3 |
|
M6 |
3- CH3 |
M18 |
5NO2 |
|
M7 |
4-OH |
M19 |
4-NH(CH3)2 |
|
M8 |
4-CH3 |
M20 |
4-OCH3 |
|
M9 |
4-OH, 5-CH3 |
M21 |
4-NH2 |
|
M10 |
5-CH3 |
M22 |
4,5-(OH)2 |
|
M11 |
6-OCH3 |
M23 |
5-OH |
|
M12 |
5-OCH3, 5-OH |
M24 |
5- CH3 |
2.2.2 Protein Preparation
The three-dimensional crystal structure of the target protein α-glucosidase (PDB ID: 3A4A) was retrieved from the RCSB Protein Data Bank. The selected protein structure contained the protein backbone, with the water molecules, co-crystallized ligands, and other heteroatoms deleted. The atoms of the hydrogen were inserted, and the protein structure was optimized for good geometry for further molecular docking studies [15].
2.2.3 Molecular Docking
Molecular docking analysis was carried out using ArgusLab 4.0. It was concluded that the prepared protein is the receptor protein, while the optimized ligands are the input molecules. It was picked by using the active site residues or the position of the co-crystallized molecule in the binding site. Docking was done with the algorithm, and numerous poses were generated for each of the ligands [16]. Binding energy scores and interaction profiles were used to assess the docked conformations. The binding pose of each ligand was determined based on the lowest binding energy and the favorable interaction with the amino acids, including hydrogen bonds, hydrophobic interactions, and van der Waals forces.
2.2.4 Molecular visualization
Three-dimensional (3D) visualization of docked ligand-protein complexes was done using Discovery Studio Visualizer (BIOVIA). 3D images of the docked complexes were generated by the software, and the orientation of the ligands in the active binding pocket of the model protein was analyzed [17].
2.3 In Silico ADMET and Drug Likeness Prediction
Pharmacokinetic properties and drug-likeness profiles of the designed ligands (M1-M24) were analyzed utilizing online computational tools. The chemical structure of selected designed compounds was drawn using the software ChemDraw and converted to canonical SMILES format, entered as input for the ADME analysis. The Absorption, Distribution, Metabolism, and Excretion (ADME) properties of the ligands were predicted with the aid of the SwissADME web server. Physicochemical and pharmacokinetic properties such as molecular weight, LogP, aqueous solubility, gastrointestinal (GI) absorption, blood-brain barrier (BBB) permeation, bioavailability scores, and synthetic accessibility were investigated for the evaluation of drug-likeness of the compounds. The toxicity profiles of the designed ligands were predicted by using ProTox-II. The SMILES of every compound were submitted to determine such parameters as LD50 values and toxicity classes. The drug likeness of the compounds was evaluated using Molsoft. Drug-likeness scores and biological activity predictions were then performed based on the structural features using the Molsoft platform, giving insight into the potential interaction of the compounds with biological factors [18].
3. RESULTS AND DISCUSSION
3.1 Molecular Docking Study
Molecular docking was subsequently carried out to understand the binding affinity of the designed 1-(substituted phenyl)-N-(2-(2-(pyridin-2-yloxy) ethoxy) ethyl) methanimine derivatives (M1–M24) in the active site of α-glucosidase. Docking results are summarized and displayed as a table including the binding affinity, hydrogen bond interactions, bond length, and interacting amino acid residues (Table 1). The clinically-used α-glucosidase inhibitor, Miglitol (-7.55 kcal/mol), docked in this receptor and made interactions through six hydrogen bonds with the following residues: ILE272, SER298, GLU296, ASN259, HIS295, and ALA295, which are present in the receptor. All of the designed derivatives showed a docking score from -8.30 to -10.96 kcal/mol, with improved docking scores as compared to Miglitol. Among the evaluated compounds, M1 exhibited the highest binding affinity with a docking score of -10.96 kcal/mol, followed by M14 (-10.43 kcal/mol), M6 (-10.40 kcal/mol), M22 (-10.20 kcal/mol), M9 (-10.06 kcal/mol), M2 (-10.03 kcal/mol), M10 (-10.02 kcal/mol), and M24 (-10.01 kcal/mol). The obtained data indicate that the designed methanimine derivatives exhibit very good properties for binding into the catalytic pocket of α-glucosidase.
Analysis of hydrogen bonding interactions indicated that several polar interactions with important residues at the active site of the docked complexes were responsible for the stability of the complexes. The five H-bond interactions between compound M1 and GLN279, ARG315, ARG442, and ASN415, and the six H-bond interactions between compound M22 and HIS112, GLN182, ARG442, and ASP69 resulted in very stable complexes with the enzyme, suggesting that these would be excellent candidates for further development. The same was obtained for M9, as it interacted with five hydrogen bonds with the residues TYR347, ARG442, ASP69, and THR306, while M14 interacted through four hydrogen bonds with the residues ARG442, ARG213, GLN353, and THR306. Compound M12 was also observed to have six hydrogen bonds with GLU405, TYR416, and ASN417, but its docking score of (-9.03 kcal/mol) was not as high as the top-ranked compounds, indicating that binding affinity is dependent on both hydrogen bonding and hydrophobic interactions.
The hydrogen bond distances observed ranged from 2.22 to 3.00 Å, which were held to be strong and energetically favorable interactions between the active site of the enzyme and the interacting ligands. Several amino acid residues like ARG442, GLN279, THR306, ASP69, GLN353, ARG315, TYR347, and ASN415 repeatedly participated in the interactions with the ligand and were observed to play a crucial role in molecular recognition and stabilization of the docked complexes. The ARG442 residue was present in the highest number of residues interacting with the majority of high-scoring derivatives, indicating that ARG442 plays a vital role in the binding of the ligand in the active pocket of α-glucosidase. Interestingly, compound M8 yielded a docking score of -9.80 kcal/mol and did not showcase any hydrogen-bond interaction, suggesting that it primarily interacts by hydrophobic, van der Waals, and π-related interactions. The observations confirm that hydrogen bonding is not the only factor involved in binding affinity and that non-covalent complementary interactions play a major role in the overall stability of the protein–ligand association.
The overall docking results suggest that the designed 1-(substituted phenyl)-N-(2-(2-(pyridin-2-yloxy) ethoxy) ethyl) methanimine derivatives have very good binding affinity towards α-glucosidase with a higher dock score compared to the reference compound Miglitol. The compounds M1, M14, M6, M22, M9, M2, M10, and M24 were found to be the most promising candidates, exhibiting promising binding energies and interactions with the key catalytic residues. The three-dimensional molecular visualization of the docked protein-ligand complexes, as shown in Figure 1, shows the binding orientation of the selected ligands in the active site of α-glucosidase, emphasizing their good orientation within the catalytic pocket. The complexes show that the stabilizing interactions of the ligands involve mostly hydrogen bonds, hydrophobic contacts, π-π stacking, and van der Waals interactions between the ligand and key residues in the active site. These interactions facilitate the excellent docking scores found for the lead compounds and reveal structural details of how they might inhibit α-glucosidase. Based on these results, the authors believe that the derivatives are promising candidates for further studies by molecular dynamics simulation, chemical synthesis, and biological testing in order to confirm their α-glucosidase inhibitory bioactivity as possible antidiabetic agents.
|
Compound ID |
Docking score |
No. of H -bonds |
Amino acid Interaction |
|
Miglitol |
-7.5509 |
6 |
272ILE, 298SER, 296GLU, 259ASN, 295HIS, 295ALA |
|
M1 |
-10.9634 |
5 |
279GLN, 442ARG, 442ARG, 415ASN, 315ARG |
|
M2 |
-10.0344 |
2 |
279GLN, 315ARG |
|
M3 |
-9.5263 |
1 |
277GLU |
|
M4 |
-9.7679 |
1 |
277GLU |
|
M5 |
-9.1068 |
1 |
321PHE |
|
M6 |
-10.3959 |
2 |
442ARG,442ARG |
|
M7 |
-9.2499 |
5 |
240SER, 242ASP, 279GLN, 69ASP, 442ARG |
|
M8 |
-9.7969 |
0 |
- |
|
M9 |
-10.064 |
5 |
347TYR, 442ARG, 442ARG, 69ASP, 306THR |
|
M10 |
-10.015 |
1 |
156LYS |
|
M11 |
-9.0329 |
2 |
315SRG, 442ARG |
|
M12 |
-9.0307 |
6 |
405GLU, 405GLU, 416TYR, 417ASN, 416TYR, 416TYR |
|
M13 |
-9.4847 |
4 |
442ARG, 353GLN, 213ARG, 306THR |
|
M14 |
-10.425 |
4 |
442ARG, 213ARG, 353GLN, 306THR |
|
M15 |
-9.5331 |
3 |
69ASP, 307ASP, 442ARG |
|
M16 |
-9.5212 |
4 |
353GLN, 306THR, 442ARG, 442ARG |
|
M17 |
-9.5830 |
4 |
415ASN, 433PHE, 317ASN, 423HIS |
|
M18 |
-9.2830 |
4 |
279GLN, 442ARG, 442ARG, 69ASP |
|
M19 |
-8.2969 |
3 |
242ASP, 280HIS, 279GLN |
|
M20 |
-9.3859 |
4 |
347TYR, 350ASN, 442ARG, 411GLU |
|
M21 |
-8.4559 |
4 |
280HIS, 306THR, 315ARG, 279GLN |
|
M22 |
-10.1993 |
6 |
112HIS, 182GLN, 442ARG, 69ASP, 182GLN, 182GLN |
|
M23 |
-9.2233 |
4 |
317ASN, 423HIS, 235ASN, 235ASN |
|
M24 |
-10.0135 |
1 |
156LYS |
Table 1. Molecular docking simulation of the novel designed 1-(substituted phenyl)-N-(2-(2-(pyridin-2-yloxy) ethoxy) ethyl) methanimine derivatives.
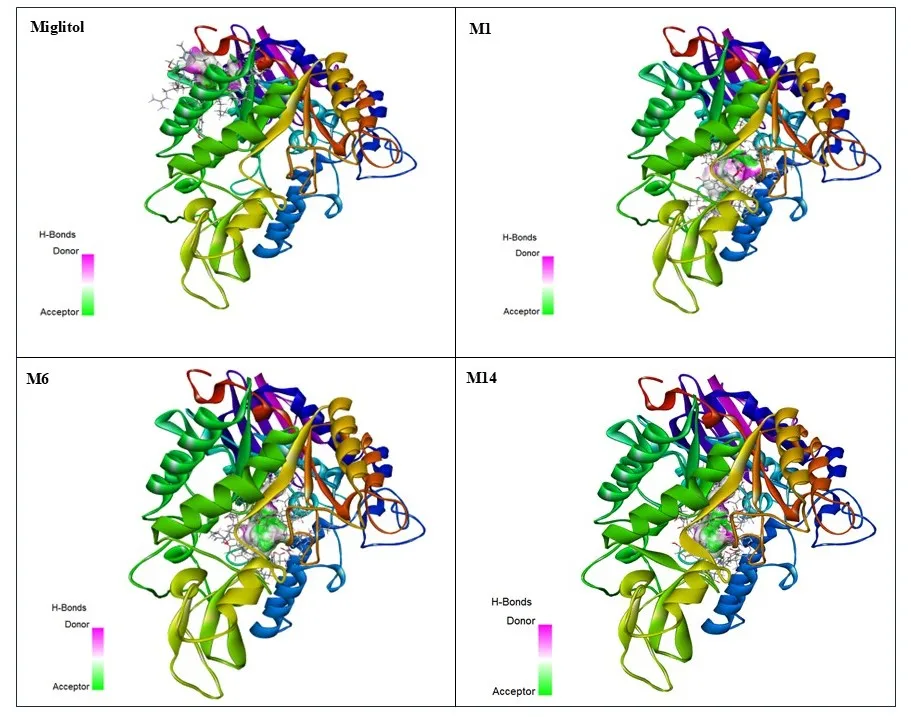
Figure 1. 3D Three-dimensional molecular docking visualization of the protein-ligand complexes of Miglitol and the potent designed derivatives bound to the active site of α-glucosidase (PDB ID: 3A4A).
3.2 In Silico Pharmacokinetic Study and Drug-Likeness Evaluation
The pharmacokinetic characteristics and drug-likeness of the designed derivatives (M1–M24) were evaluated using in silico ADMET prediction tools to assess their suitability as potential drug candidates. All the calculated physicochemical and pharmacokinetic parameters (molecular weight (MW), number of heavy atoms (nHA), number of aromatic heavy atoms (nAHA), number of rotatable bonds (nRB), number of hydrogen bond acceptor (nHBA), gastrointestinal absorption (GIT), blood–brain barrier (BBB) permeability, number of hydrogen bond donor (nHBD), drug-likeness score, and predicted acute oral toxicity (LD50)) are summarized in Table 2.
Molecular weights of designed derivatives were in the range of 270.33 – 430.67 g/mol, which is below the generally accepted upper limit of 500 g/mol advised by Lipinski's rule of five. The M1 had the lowest molecular weight of 270.33 g/mol, while the M7 had the highest molecular weight of 430.67 g/mol. Counties of 20 to 25 heavy atoms were observed between the derivatives, and the heavy atoms in the aromatic ring were levied at 12 atoms for each derivative, thus maintaining a common framework for the aromatic system throughout the designed series. The number of rotatable bonds of structural flexibility was varied between eight and 10, indicating moderate flexibility of the molecule, which is desirable for good receptor binding and not causing molecular instability. Hydrogen bond acceptors varied from 4 to 7, while hydrogen bond donors showed ranges between 0 and 2, which supports all the derivatives having suitable properties for favorable oral bioavailability. Twenty-four derivatives were predicted with gastrointestinal absorption, and it was found that all of them have high GIT absorption, suggesting their potential to be highly absorbed by the oral route and thus negligible side effects in the nervous system. system targets or were avoided for peripherally acting drugs to avoid neurological side effects. Predicted drug/likeness scores ranged from -0.59 to 0.45. The results tended to be more positive for the drug likeness for most of the compounds, suggesting a good physicochemical property for drug development. The compound with the highest drug-likeness score was M7 (0.45), followed by M13 (0.43) and M9 (0.42), indicating an optimal combination of molecular features and pharmacokinetic properties. However, compounds M5 (-0.59), M11 (-0.24), M1 (-0.14), M19 (-0.07), and M8 (-0.03) had slightly negative scores and may also not preclude further consideration for biological evaluation, especially in the presence of encouraging biological activity. Acute oral toxicity prediction revealed good safety profiles of the molecules designed. The acute toxicity estimates ranged from 1250 to 2000 mg/kg estimated LD50, indicating relatively low toxicity.
The highest predicted LD50 values were obtained for compounds M3, M9, M11, M12, M14-M17, and M23, with 2000 mg/kg, which are comparatively less toxic than derivatives with 1250 mg/kg predicted LD50 values. In conclusion, the in silico pharmacokinetic evaluation showed that the engineered derivatives have drug-like attributes like low toxicity in general, good gastrointestinal absorption, acceptable toxicity, molecular flexibility, adequate hydrogen-bonding ability, and acceptable molecular weight. Most compounds also presented good drug-likeness scores, while showing BBB permeability, suggesting they could be orally active lead compounds. M7, M9, M12, and M13 showed a very well-balanced pharmacokinetic profile – these derivatives absorbed best into the gut, had good BBB permeability, good drug-likeness scores, and acceptable toxicity predictions. All this, along with their biological activity and molecular docking results confirm that these compounds could be used for experimental investigation of pharmacological aspects in the further steps of their optimization.
|
Compound ID |
MW (g/mol) |
nHA |
nAHA |
nRB |
nHBA |
nHBD |
GIT |
Drug likeness |
LD 50 (mg/kg) |
|
M1 |
270.33 |
20 |
12 |
8 |
4 |
0 |
High |
1.14 |
1250 |
|
M2 |
304.77 |
21 |
12 |
8 |
4 |
0 |
High |
0.27 |
1250 |
|
M3 |
319.79 |
22 |
12 |
8 |
4 |
1 |
High |
0.19 |
2000 |
|
M4 |
322.76 |
22 |
12 |
8 |
5 |
0 |
High |
0.28 |
1250 |
|
M5 |
394.22 |
24 |
12 |
9 |
6 |
0 |
High |
0.59 |
1250 |
|
M6 |
396.22 |
21 |
12 |
8 |
4 |
0 |
High |
1.16 |
1250 |
|
M7 |
430.67 |
22 |
12 |
8 |
4 |
0 |
High |
0.45 |
1250 |
|
M8 |
284.35 |
21 |
12 |
8 |
4 |
0 |
High |
0.03 |
1250 |
|
M9 |
300.35 |
22 |
12 |
8 |
5 |
1 |
High |
0.42 |
2000 |
|
M10 |
284.35 |
21 |
12 |
8 |
4 |
0 |
High |
0.16 |
1250 |
|
M11 |
300.35 |
22 |
12 |
9 |
5 |
0 |
High |
0.24 |
2000 |
|
M12 |
316.35 |
23 |
12 |
9 |
6 |
1 |
High |
0.29 |
2000 |
|
M13 |
395.25 |
24 |
12 |
9 |
6 |
1 |
High |
0.43 |
1250 |
|
M14 |
328.36 |
24 |
12 |
9 |
6 |
1 |
High |
1.09 |
2000 |
|
M15 |
365.22 |
22 |
12 |
8 |
5 |
1 |
High |
1.09 |
2000 |
|
M16 |
286.33 |
21 |
12 |
8 |
5 |
1 |
High |
0.87 |
2000 |
|
M17 |
345.35 |
25 |
12 |
10 |
7 |
0 |
High |
0.98 |
2000 |
|
M18 |
315.32 |
23 |
12 |
9 |
6 |
0 |
High |
1.29 |
1250 |
|
M19 |
313.39 |
23 |
12 |
9 |
4 |
0 |
High |
0.07 |
1250 |
|
M20 |
300.35 |
22 |
12 |
9 |
5 |
0 |
High |
0.29 |
1250 |
|
M21 |
285.34 |
21 |
12 |
8 |
4 |
1 |
High |
0.12 |
1250 |
|
M22 |
302.33 |
22 |
12 |
8 |
6 |
2 |
High |
0.29 |
1250 |
|
M23 |
286.33 |
21 |
12 |
8 |
5 |
1 |
High |
0.42 |
2000 |
|
M24 |
284.35 |
21 |
12 |
8 |
4 |
0 |
High |
0.16 |
1250 |
MW: Molecular weight, nHA: No. of heavy atom, nAHA: No. of Aromatic heavy atoms, nRB: No. of Rotatable bonds nHBA: No of H-bond acceptors, nHBD: No. of H-bond donor, LD: Lethal dose.
Table 2. In silico ADMET and physicochemical profiles of the designed 1-(substituted phenyl)-N-(2-(2-(pyridin-2-yloxy) ethoxy) ethyl) methanimine derivatives.
CONCLUSION
In the present study, novel N-(2-(2-(pyridin-2-yloxy) ethoxy) ethyl) methanimines, also known as 1-(substituted phenyl) derivatives, were revealed as potential α-glucosidase inhibitors. The designed compounds exhibited high binding affinity levels and had more favorable binding interactions with the crucial catalytic residues than did Miglitol. ADMET analysis also showed good drug-like properties, high gastrointestinal absorption, and minimal predicted toxicity. Derivatives of E17 were evaluated, with M1, M6, M14, and M22 showing the most promising lead molecules. These findings offer a robust computational basis for their chemical synthesis, experimental α-glucosidase inhibition studies, and subsequent lead optimization for the development of novel antidiabetic agents.
Acknowledgments
We are very thankful to our institute, the Kamla Institute of Pharmaceutical Sciences, Bhilai, Chhattisgarh, India, and the University, Shri Shankaracharya Professional University, Bhilai, Chhattisgarh, India, for providing us with facilities and allowing us to conduct the research work.
REFERENCES

Anju Daharia*, Mousmi Sahu, Pooja Verma, Molecular Docking And Pharmacokinetic Evaluation Of Novel 1-(Substituted Phenyl)-N-(2-(2-(Pyridin-2-Yloxy) Ethoxy) Ethyl) Methanimine Derivatives As Potential Α-Glucosidase Inhibitors, Int. J. Sci. R. Tech., 2026, 3 (7), 190-199. https://doi.org/10.5281/zenodo.21261704
 10.5281/zenodo.21261704
10.5281/zenodo.21261704