
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Shri Shankaracharya professional University, Bhilai, Chhattisgarh, India 490020
Objective: The aim of the present study was to design and evaluate a new series of benzopyran derivatives based on ester as possible nephroprotective agents using molecular docking studies and in silico ADMET analysis. The designed compounds were developed by structural modifications of the flavonol skeleton, from the known nephroprotective and antioxidant effects of quercetin. Materials and Methods: A novel series of 10 ester-functionalized chromone derivatives namely (3a-3j) were designed on the basis of 3-hydroxy-2-phenyl-4H-chromen-4-one scaffold, which were docked with its target protein (PDB ID: 2AZ5) obtained from the kidney protein library using Molegro Virtual Docker (MVD v6.0). The binding affinity, molecular interactions, hydrogen-bonding pattern, and most important residues in the active site were systematically explored. Moreover, the pharmacokinetic properties and drug-likeness characteristics of the natural antioxidants were evaluated through the SwissADME and pkCSM platforms and the toxicity profile and safety parameters were predicted by the ProTox-3.0 web server. Results and Discussion: Molecular docking shows that all prepared derivatives have higher binding energies than the reference compound Quercetin (MolDock score: -106.906 kcal/mol). Synthesized candidates showed compound 3i to have the best binding affinity of -143.129 kcal/mol, making considerable interactions with the amino acid residues Lys98, Pro117 and Tyr119. Based on ADMET analysis, the pharmacokinetics of all the compounds were good, meeting Lipinski's rule of five and had high gastrointestinal absorption. There was low amount of BBB permeability seen for the derivatives, which resulted in low CNS exposure. Conclusion: Molecular docking and integrated ADMET study showed that compound 3i is a good hit with strong binding affinity and acceptable toxicological profile; good pharmacokinetic profile. Further synthesis, biological evaluation, and mechanistic studies are warranted to validate its nephroprotective potential, as suggested by these findings.
Nephrotoxicity includes a number of disorders, including acute renal failure, CKD progression, renal tubular injury, and glomerular damage. It is a critical medical concern since it causes hospitalization, higher healthcare expenses, and death worldwide.[1] Antibiotics such as aminoglycosides,[2] amphotericin B,[3] beta-lactams,[4] cephaloridine, cephaloglycin, [9] imipenem,[5] quinolones,[6] vancomycin,[4,7] and rifampin [8] are examples of classical nephrotoxins.[9] The following pathways, including NF-kB and TNF alpha, also activate pro-inflammatory cytokines, resulting in oxidative stress and cellular apoptosis, as well as kidney injury.[10] Drug-induced nephrotoxicity can affect the glomerulus, tubular epithelium, interstitial tissues, and blood vessels in the renal vascular bed. Several processes have been attributed to the development of nephropathy in these illnesses, including changes in renal hemodynamic, direct tubule toxicity, inflammation, crystal formation, rhabdomyolysis, and thrombotic microangiopathy.[11] This mechanism activates the damage recognition system, which includes toll-like receptor 4 (TLR-4) and purinoceptor type 2X7 (P2X7), resulting in the NLRP3 inflammasome activation cascade, producing severe tubulointerstitial.[12] Numerous natural and herbal compounds, such as flavonoids and alkaloids, have positive effects on nephrotoxicity. Plants having nephroprotective characteristics are an increasingly prevalent subject in both traditional and modern medicine due to their capacity to protect the kidneys from injury in a variety of ways. These plants are resistant to the nephrotoxic effects of chemicals and medications because they contain a variety of phytochemicals, including terpenoids, polyphenols, and flavonoids.[13] An increase in kidney problems around the world has prompted studies into natural medications that could supplement conventional forms of treatment by providing a safe choice with no adverse effects. The specific receptors and proteins that render a chemical nephrotoxic vary depending on the substance. This happens because the kidneys are actively.[14] Quercetin is a natural flavonoid that has antioxidant or nephroprotective properties. Reduce ROS production. Activate the Nrf2 antioxidant response element pathway. Increase the expression of antioxidant enzymes. Suppress inflammation. Prevent kidney cell death (apoptosis).[15] The benzopyran-4-one skeleton, or chromone ring structure, has been labelled a "privileged scaffold in drug discovery." The benzopyran-4-one ring structure serves as the foundation for all flavonoid compounds. The flavonoid subclasses are formed by substituent groups at positions 2, 3, 5, 6, 7, and 8.[16] Chromones (4H-1-Benzopyran-4-ones) are an important class of oxygen heterocyclic compounds with a benzo-γ-pyrone structure. These compounds have shown a wide range of biological properties including antibacterial, anticancer, anti-inflammatory, and antioxidative, and because to their low toxicity in animals, various medicines from this family have been produced, such as Chromonar, Flavoxate, and Scutellarein. [17] Acid chloride ester pharmacophores were chosen as possible inhibitors of NFKB and TNF-alpha. The design of test ligands with increased nephroprotective efficacy is inspired by the structural properties of standard drugs such as Quercetin. [18] This is where we present our study, in which we designed an ester derivative capable of lowering kidney toxicity and oxidative stress. The 3-hydroxy-2-phenyl-4H-1-Benzopyran-4-one (Flavonol) ester derivatives have higher lipophilicity than their hydroxyl flavone counterparts, allowing them to penetrate through the cell membrane during pharmacological tests. [19]
2. MATERIAL AND METHODS
Download the target protein's 3D crystal structure in PDB file format from the Protein Data Bank (RCSB, www.rcsb.org) and use the right PDB code. Based on the following parameters: ligand co-crystallized with the protein (helps pinpoint the binding site), and protein without missing parts.[20] The formed protein structure also included water molecules and cofactors. Cofactors and water molecules appeared to be excluded from a PDB file after being loaded into the MVD Version 6.0 application. Furthermore, polarization of hydrogen, protonation states, and undefined charge assignment were done via the Molegro Virtual Docker.[21]
The 2D structures of ten benzopyran ester derivates (3a-3j) (Table 1) and a reference drug (querecetin) as ligands were obtained using ChemDraw software. The 2D structure was then transformed to a 3D structure using ChemDraw ultra, and the geometry was optimized using MM2 energy minimization in order to assure that the ligand accepts low energy. [22,23]
3-(susbtsituted-1,3-dien-1-yloxy)-2-phenyl-4H-chromen-4-one
|
Compound Id |
R |
Compound Id |
R |
|
3a |
|
3f |
|
|
3b |
|
3g |
|
|
3c |
|
3h |
|
|
3d |
|
3i |
|
|
3e |
|
3j |
|
Table 1. List of novel design ester derivatives.
MVDv 6.0 was used to perform the molecular docking simulation. Accurate imported molecular structure that is, correct atom connectivity, bond ordering, and partial atomic charges is necessary for precise docking. PDB (2AZ5) files frequently contain incorrect or missing explicit hydrogen assignments, and the bond order feature is absent from the PDB file format. By using a cavity identification technique, MVD software automatically finds possible pockets for binding sites, called cavities. To automate the benchmarking process, the experimentally observed ligand position in the binding site was surrounded by a cubic grid measuring 30 x 30 x 30 x 3. The differential evolution method's search algorithm aggressively exploits cavities found by the cavity detection algorithm. To cover the whole surface of the discovered cavity binding site, ten runs were linked to each ligand with 1500 iterations and a population size of 50. The MolDock score, H-bond score, and Rerank score were used among the several scoring functions to determine the ideal position. An optimal orientation for the protein-ligand complex was assessed in each ligand docking experiment, and the hydrogen bonds were found and labelled. MVD Score, a linear combination of E-inter, was used to analyse the ligand energy. [24]
2.4 ADMET study
The ADME (absorption, distribution, metabolism and excretion) characterizations of the several 3-hydroxy-2-phenyl-4H-1-Benzopyran-4-one Ester derivatives were evaluated using multiple web servers to predict the relevant pharmacokinetic features. The In-Silico ADME screening and drug-likeness evaluation were predicted using the free web tools pkCSM (https://biosig.lab.uq.edu.au/pkcsm/) and Swiss ADME, the toxicity study performed by Protox 3.0 (https://tox.charite.de/). [25]
3. RESULT AND DISCUSSION
3.1 Molecular Docking study
Molecular docking is an optimization problem aimed at discovering the pose of a ligand molecule with the lowest potential energy. In molecular docking, searches are conducted in the coordinate space of the target binding site, and each conformation of the ligand at its target site is scored, with the highest score indicating the most likely binding mode. Table 2 depicts the MolDock score, H-bond, heavy atom, and hydrogen bond interactions of -hydroxy-2-phenyl-4H-1-benzopyran-4-one derivatives. It is noteworthy to note that the binding energy of all proposed benzopyran-4-one ester derivatives (3a-3j) varies from -106.906 to -143.129 kcal/mol. Among the ten ligands, three were shown to have the highest MolDock scores. As a result, compared to ordinary molecules, the developed compounds create a more stable complex. Ligand 3i (E)-3-[(3-(4-hydroxy-3,5-dimethoxyphenyl) acryloyl)oxy]-2-phenyl-4H-benzopyran-4-one) showed the best results and formed complexes with an active site with mole dock score -143.129 kcal/mol and a binding site made up of amino acid residues like Try119, Pro117, and Lys98 (Figure 1).
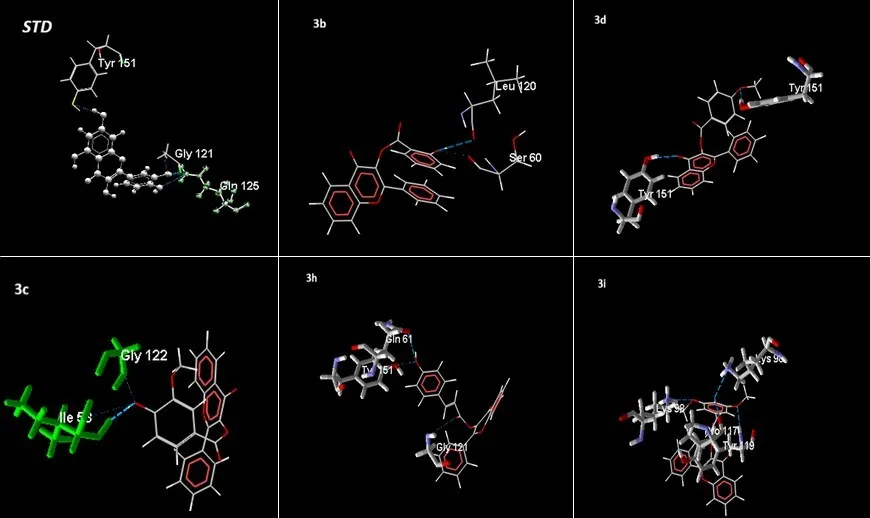
Figure 1. 2D interaction of protein-ligand complex of designed potent compounds along with standard drug against the target protein generated by the Molegro virtual docker v.6.0 software.
|
Compound ID |
MolDock Score (kcal/mol) |
H-bond (kcal/mol) |
Heavy Atoms |
H-Bond Interaction |
|
Quercetin |
-106.906 |
-8.07707 |
22 |
Tyr151, Gln125, Gly121 |
|
3a |
-115.779 |
- |
26 |
- |
|
3b |
-123.7007 |
-2.52501 |
27 |
Leu120, Ser60 |
|
3c |
-131.737 |
-3.67661 |
29 |
Ile58, Gly122 |
|
3d |
-117.44 |
-4.25559 |
28 |
Tyr151 |
|
3e |
-125.765 |
-3.1332 |
29 |
Try151 |
|
3f |
-119.478 |
- |
28 |
- |
|
3g |
-135.489 |
- |
31 |
- |
|
3h |
-136.272 |
-3.3674 |
29 |
Try151, Gly121, Gln61 |
|
3i |
-143.129 |
-9.9329 |
33 |
Try119, Pro117, Lys98 |
|
3j |
-131.235 |
- |
30 |
- |
Table: 2 The binding score of the design ligands with the target protein based on MolDock, Interaction, H-Bond, and Heavy atoms, H- Bond Interaction.
3.2 ADMET
The ADME (absorption, distribution, metabolism, and excretion) is crucial for determining how effective an inhibitor is in the body. Table 3 shows the physicochemical characteristics of the proposed inhibitors, including molecular weight, rotatable bonds, molecular formula, H-bond acceptor, H-bond donor, etc. The drug's lipophilicity indicates how well it dissolves in lipids and nonpolar liquids. The efficiency of absorption through the cell membrane is greatly dependent on this characteristic. This implies that the suggested substance will guarantee high bioavailability and is acceptable due to its superior absorption capacity. Using Swiss ADME, the compounds' drug-likeness was anticipated. According to Lipinski's Rule of Five, a medication or inhibitor may have a molecular weight (MW) of less than 500 Da, n HBA of no more than 10, n HBD of no more than 5, and TPSA of no more than 140 Ų. It is evident that the proposed ligands (3a-3j) do not violate the drug-likeness characteristics of the medications listed in Table 4. The initial stage of oral bioavailability is influenced by the drug-likeness attribute, which also affects water solubility and gastrointestinal absorption. The distribution and absorption of the chemicals were proposed by the gut and BBB. A material is considered to have weak absorption ability if its absorbance is less than 30%. According to reports, the experimental value of the developed ligands' absorption was higher for GI absorption, suggesting that the drug was acceptable. The blood-brain barrier (BBB) protects the brain from any undesirable substances and either increases the efficacy of medications or reduces their adverse effects. chemicals with log BB larger than 0.3 are supposed to easily cross the blood-brain barrier, while chemicals with BB less than -1 are thought to disperse poorly in the brain. According to the computed values, the ligand values are less than -1, meaning that the ligand's active site is not for the central nervous system. The log PS value, which must be smaller than -3 in accordance with the literature, provides additional evidence of the intended ligand's non-CNS activity. Renal clearance leads to the drug's clearance, which is connected to its bioavailability.
ProTox-3.0 and ADMETLab 3.0 were used to evaluate the toxicological profile of these developed (3a-3j) compounds. The compounds have good LD50 values, indicating moderate acute toxicity, as indicated by the predicted toxicity classes falling between IV and V. These molecules' accessibility was also assessed, and the results showed that their scores ranged from 3.36 to 3.96, indicating that their synthetic feasibility is also acceptable. According to estimates, the nephrotoxicity of these developed compounds ranges from 0.06 to 0.383, which is far below the threshold value of 0.0-1.0, indicating minimal nephrotoxic effects. Overall, these developed compounds have demonstrated good pharmacokinetics and safety features based on all of these In-Silico investigations.
|
Compound ID |
Molecular Formula |
MW (g/mol) |
Lipinski rule |
nHBA |
nHBD |
TPSA |
|
3a |
C22H14O4 |
342.34 |
Yes |
4 |
0 |
56.51 |
|
3b |
C22H14 O5 |
358.08 |
Yes |
5 |
1 |
76.74 |
|
3c |
C23H16 O6 |
388.09 |
Yes |
6 |
1 |
85.97 |
|
3d |
C23H16 O5 |
372.10 |
Yes |
5 |
0 |
65.74 |
|
3e |
C22H14 O7 |
390.07 |
Yes |
7 |
3 |
117.20 |
|
3f |
C24H16 O4 |
368.10 |
Yes |
4 |
0 |
56.51 |
|
3g |
C25H18 O6 |
414.11 |
Yes |
6 |
1 |
85.97 |
|
3h |
C24H16 O5 |
384.10 |
Yes |
5 |
1 |
76.74 |
|
3i |
C26H20O7 |
444.12 |
Yes |
7 |
1 |
95.20 |
|
3j |
C24H16 O6 |
400.09 |
Yes |
6 |
2 |
96.97 |
Table: 3 Physiochemical characteristics and drug-likeness properties of the test ligands
|
Compound ID |
GI Absorption |
BBB |
CNS |
Predicted toxicity class |
ETC |
Nephrotoxicity |
Synthetic associability |
|
3a |
High |
0.046 |
-1.655 |
4 |
0.726 |
0.067 |
3.36 |
|
3b |
High |
-0.105 |
-1.843 |
4 |
0.644 |
0.079 |
3.44 |
|
3c |
High |
-0.568 |
-2.085 |
4 |
0.576 |
0.104 |
3.62 |
|
3d |
High |
-0.091 |
-1.866 |
4 |
0.687 |
0.156 |
3.51 |
|
3e |
High |
-1.411 |
-3.416 |
4 |
0.355 |
0.006 |
3.52 |
|
3f |
High |
-0.099 |
-1.617 |
5 |
0.784 |
0.383 |
3.60 |
|
3g |
High |
-0.645 |
-1.969 |
5 |
0.616 |
0.302 |
3.79 |
|
3h |
High |
-0.422 |
-1.804 |
5 |
0.657 |
0.244 |
3.62 |
|
3i |
High |
-0.804 |
-2.879 |
5 |
0.588 |
0.309 |
3.96 |
|
3j |
High |
-0.562 |
-2.062 |
5 |
0.521 |
0.086 |
3.67 |
Table: 4 Absorption, distribution, excretion, and toxicity of the test ligands.
CONCLUSION
The present work confirmed that novel Benzopyran derivatives carrying an ester functionality could be good nephroprotective agents by using an inbuilt computational approach. Molecular docking studies proved interesting interactions occurred between the designed compound and the selected target protein and further modification of the benzopyran scaffold by incorporating an ester group could lead to better binding at the target site and molecular recognition. The interaction profiles indicated the ability of these derivatives to form stable protein-ligand complexes within the active site. Moreover, the ADMET parameters indicated good drug-like properties, favourable pharmacokinetic properties, and a safety profile for the designed molecules. The compounds were found to be compliant with drug-likeness parameters and were potentially promising for further drug development studies. In conclusion, the results suggest that ester derivatives of benzopyrans are an interesting group of compounds for the identification of novel nephroprotective drugs. The study offers a solid computational basis to drive any future synthetic application and study of the biological activity. Experimental validation of the molecules through synthesis, characterization, in vitro nephroprotective screening, mechanistic studies, and in vivo evaluation is further key to confirm their therapeutic potential and progress toward developing effective nephroprotective drugs.
REFERENCES

Swapnil Deshmukh, Vaibhav Tiwari*, In Silico Design Of Novel Ester-Based Derivatives As Nephroprotective Potential: Molecular Docking And Pharmacokinetic Evaluation, Int. J. Sci. R. Tech., 2026, 3 (6), 1401-1408. https://doi.org/10.5281/zenodo.20840071
 10.5281/zenodo.20840071
10.5281/zenodo.20840071