
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
K. V. N. Naik S. P. Sanstha’s, Institute of Pharmaceutical Education & Research, Nashik, 422002, Maharashtra, India.
Invasive fungal infections continue to impose a substantial global health burden, with treatment further complicated by the emergence of resistance to conventional antifungal agents. The tubulin protein complex, specifically the colchicine-binding site of the heterodimeric alpha-beta tubulin, represents a validated and pharmacologically tractable molecular target in fungal pathogens. In the present study, eight novel substituted benzimidazole-Schiff base hydrazone derivatives were rationally designed and subjected to comprehensive in silico evaluation comprising molecular docking, ADME prediction, and toxicological profiling. The crystal structure of the tubulin-colchicine stathmin-like domain complex (PDB: 1SA0, resolution 3.58 Å) was employed as the receptor for docking simulations performed using PyRx with AutoDock Vina algorithms. Binding affinity scores for the designed molecules ranged from -7.0 to -9.0 kcal/mol. Molecule 3 (C??H??IN?; MW 452.29 g/mol), bearing a 4-iodo substituent on the terminal phenyl ring and a 4-methyl group on the central aryl moiety, exhibited the highest binding affinity of -9.0 kcal/mol, surpassing the reference drug mebendazole (-8.7 kcal/mol). Key binding interactions of Molecule 3 with residues Lys352, Val236, Ala314, and Phe255 included hydrogen bonding, halogen interaction mediated by the iodo group, and aromatic pi-pi stacking. Drug-likeness assessment according to Lipinski, Veber, and Egan criteria confirmed that the majority of the designed molecules fulfilled oral bioavailability requirements. Pharmacokinetic profiling predicted high gastrointestinal absorption for Molecules 1, 3, and 5. ProTox-II toxicological analysis assigned most compounds to Toxicity Class 4, indicating a comparatively safer acute oral toxicity profile relative to mebendazole, which was categorised as Class 2. These computational findings collectively identify Molecule 3 as a promising lead candidate warranting further experimental investigation for antifungal drug development.
Fungal infections have emerged as a critical area of unmet medical need, particularly among immunocompromised populations including organ transplant recipients, cancer patients undergoing chemotherapy, individuals with HIV/AIDS, and those on prolonged corticosteroid therapy [1]. The spectrum of clinically significant fungal pathogens encompasses species of Candida, Aspergillus, Cryptococcus, and dermatophytic fungi, collectively responsible for an estimated 1.5 million deaths annually worldwide [2]. Despite this considerable disease burden, the antifungal drug armamentarium remains limited to a handful of mechanistic classes, namely the azoles targeting ergosterol biosynthesis, polyenes disrupting membrane integrity, echinocandins inhibiting beta-glucan synthesis, and antimetabolites such as flucytosine [3]. This narrow therapeutic landscape is further constrained by intrinsic and acquired resistance mechanisms that have progressively eroded the clinical utility of frontline agents. Azole resistance in Candida glabrata and Candida auris, along with the emergence of multi-azole- resistant Aspergillus fumigatus strains, underscores the urgency for new antifungal scaffolds acting through alternative molecular mechanisms [4].
Tubulin, the structural protein component of microtubules, has long been recognised as a selective antifungal target, primarily because of meaningful differences in the colchicine-binding site between fungal and mammalian tubulin isoforms [5]. Microtubules are dynamic polymers formed by the polymerisation of alpha-beta tubulin heterodimers and are indispensable for mitotic spindle assembly, chromosome segregation, and intracellular transport [6]. Agents that bind the colchicine site stabilise the curved conformation of the tubulin heterodimer, thereby preventing longitudinal contacts required for polymerisation [7]. Benzimidazole antifungals such as mebendazole, albendazole, and thiabendazole exploit this mechanism, binding selectively to fungal tubulin owing to structural divergences at residues that form the colchicine pocket, thereby achieving antiparasitic and antifungal activity at doses that spare mammalian microtubules to a meaningful extent [8]. However, the pharmacokinetic limitations of classical benzimidazoles, including poor aqueous solubility, erratic gastrointestinal absorption, and systemic adverse effects, highlight the need for structurally optimised analogues.
Benzimidazole is a privileged pharmacophore in medicinal chemistry, incorporating a benzene ring fused to an imidazole nucleus to create a bicyclic aromatic system capable of extensive hydrogen bonding, aromatic interactions, and electronic delocalisation [9]. The scaffold has been exploited across a broad range of biological activities including antifungal, antiparasitic, antiviral, anticancer, and anti-inflammatory applications [10]. Structural modifications at the N-1 position, C-2 position, and through Schiff base condensation with the hydrazinyl function have been demonstrated to substantially modulate binding affinity, selectivity, and physicochemical properties [11]. The introduction of hydrazone linkages connects the benzimidazole core to a substituted terminal aromatic ring, creating a conjugated framework that increases planarity and enhances pi-stacking capacity within binding pockets [12].
Computational drug discovery approaches have transformed the efficiency and directionality of the lead identification phase [13]. Molecular docking provides a mechanistic understanding of ligand-protein complementarity at the atomic level, while in silico ADME prediction and toxicity profiling through tools such as SwissADME and ProTox-II allow for the rapid elimination of pharmacokinetically inferior or potentially toxic candidates at early stages of the discovery pipeline [14,15]. These computational filters are invaluable for prioritising compounds for synthesis and experimental validation, significantly reducing both cost and time-to-candidate [16].
Given these considerations, the present study was designed to rationally construct a series of eight benzimidazole-based Schiff base hydrazone derivatives carrying diverse substituents, including bromo, fluoro, iodo, nitro, trifluoromethyl, hydrazinyl, cyano, and carboxylic acid groups, on both the central phenyl aryl ring and the terminal aromatic moiety. These compounds were then evaluated computationally using the 1SA0 crystal structure of the tubulin-colchicine stathmin-like domain complex as the molecular target. The objectives were to determine binding affinities relative to mebendazole, analyse structure-activity relationships at the electronic and steric level, assess oral drug-likeness according to established pharmacokinetic rules, and evaluate the preliminary toxicological risk profile, collectively providing a rational computational basis for the advancement of optimised benzimidazole antifungal leads.
2. MATERIALS AND METHODS
2.1 Target Protein Selection and Preparation
The crystal structure of the tubulin-colchicine stathmin-like domain complex (PDB ID: 1SA0) was retrieved from the RCSB Protein Data Bank (https://www.rcsb.org). This structure, resolved at 3.58 Å by X-ray crystallography, contains the alpha-beta tubulin heterodimer bound to colchicine within the interdimer interface, representing the classical colchicine-binding site that is targeted by benzimidazole antifungals [7]. The protein was imported into BIOVIA Discovery Studio Visualizer 2021 for structural inspection, removal of crystallographic water molecules, co-crystallised ligands, and ionic cofactors not essential to binding pocket architecture. Polar hydrogen atoms were added, and Gasteiger partial charges were assigned to all protein atoms to ensure accurate electrostatic representation during the docking calculation. Energy minimisation of the prepared protein was performed to relieve steric clashes arising from crystal packing forces prior to grid box generation.
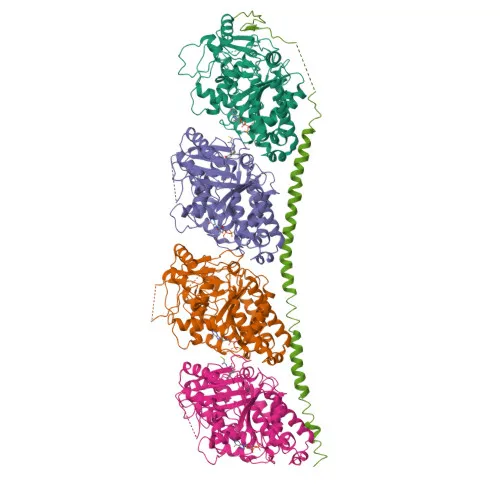
Figure 1. Three-dimensional structure of the tubulin-colchicine stathmin-like domain complex (PDB ID: 1SA0) used for molecular docking studies.
2.2 Ligand Design and Structure Preparation
The eight target molecules were designed as substituted benzimidazole-Schiff base hydrazone conjugates featuring a 2-arylbenzimidazole core coupled through a hydrazone linkage (C=N-NH) to a variably substituted terminal aryl group. Structural diversity was introduced by positioning different substituents on both the central aryl ring attached to the C-2 position of benzimidazole and on the terminal phenyl ring of the hydrazone moiety. Specifically, the central ring carried either a 4-methyl, 5-nitro, 5-trifluoromethyl, 5-hydrazinyl, or 5-carboxylic acid group, while the terminal ring featured 4-bromo, 3-bromo, 4-iodo, 3-fluoro, 4-fluoro, 3-chloro, 4-cyano, or 3-iodo substituents. Two-dimensional chemical structures were drawn using ACD/ChemSketch (Advanced Chemistry Development, Inc.), and canonical SMILES strings were generated for each compound. Structures were subsequently imported into PyRx 0.8 (Virtual Screening Software) where they were converted to three-dimensional format using the integrated Open Babel toolbox. Each ligand underwent geometry optimisation using the Universal Force Field (UFF), followed by energy minimisation to achieve the lowest-energy three-dimensional conformation, which was then converted to the PDBQT format required for AutoDock Vina docking.
Mebendazole (Câ‚₆Hâ‚₃N₃O₃; MW 295.29 g/mol), the WHO-listed benzimidazole reference antifungal drug, was retrieved from the PubChem database and subjected to identical preparation procedures to serve as the positive control.
2.3 Molecular Docking Procedure
All docking simulations were carried out using AutoDock Vina as implemented in PyRx 0.8, which employs an iterated local search global optimiser combined with a knowledge-based empirical scoring function to estimate binding free energies in kcal/mol [13]. A grid box was defined around the colchicine-binding site of 1SA0 at coordinates corresponding to the centroid of the co-crystallised colchicine molecule. The grid dimensions were set at 25 x 25 x 25 Å with a spacing of 0.375 Å to adequately encompass the binding pocket and accommodate the planned molecular scaffolds. The exhaustiveness parameter was set to 8, generating nine binding modes per ligand from which the conformation displaying the lowest binding free energy was selected as the representative docked pose. All docking results were validated by re-docking colchicine into the binding site and verifying that the re-docked pose reproduced the crystallographic binding conformation within an acceptable root-mean-square deviation threshold.
2.4 Interaction Analysis
Detailed analysis of non-covalent interactions between each docked ligand and the binding site residues was performed using BIOVIA Discovery Studio Visualizer. Two-dimensional ligand-protein interaction diagrams were generated to identify hydrogen bond donors and acceptors, hydrophobic contacts, pi-pi stacking interactions, halogen bonds, and electrostatic interactions. Three-dimensional binding poses were visualised to assess the spatial orientation of each ligand within the active site cavity and to understand the structural basis for observed differences in binding affinity across the series.
2.5 ADME and Drug-Likeness Prediction
Pharmacokinetic and physicochemical properties were predicted for all eight designed molecules and mebendazole using the SwissADME web server (http://www.swissadme.ch) [14]. Input was provided as canonical SMILES strings. Parameters evaluated included molecular weight, topological polar surface area (TPSA), number of hydrogen bond acceptors and donors, count of rotatable bonds, lipophilicity (consensus log P), gastrointestinal (GI) absorption, blood-brain barrier (BBB) permeation, and ESOL-based aqueous solubility class. Drug-likeness was evaluated against the Lipinski Rule of Five [17], Veber criteria [18], and Egan filter [19]. Bioavailability score was calculated from the radar plot parameters provided by SwissADME. Medicinal chemistry filters including PAINS (Pan Assay Interference Compounds) and Brenk structural alerts were applied to identify potentially promiscuous or reactive substructures [14].
2.6 Toxicity Prediction
In silico toxicological profiling was conducted using the ProTox-II webserver (https://tox-new.charite.de/protox_II/) [15], which employs machine learning models trained on data from established toxicological databases to predict acute oral toxicity LDâ‚…â‚€ values and GHS toxicity classification. Additional endpoint predictions were obtained for hepatotoxicity, nephrotoxicity, neurotoxicity, respiratory toxicity, cardiotoxicity, carcinogenicity, immunotoxicity, mutagenicity, and cytotoxicity. Probability scores for each endpoint were recorded alongside qualitative active-inactive classifications to provide a comprehensive preliminary safety assessment for each compound.
3. RESULTS AND DISCUSSION
3.1 Molecular Design and Structural Characteristics
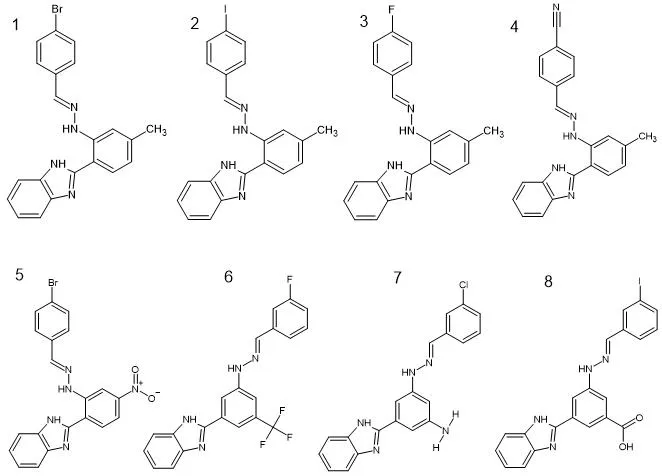
Figure 2. Chemical structures of the designed substituted benzimidazole derivatives (Molecules 1–8).
The eight benzimidazole-Schiff base hydrazone derivatives were designed based on the known pharmacophoric requirements of colchicine-site ligands, which typically require a planar aromatic system capable of occupying the hydrophobic pocket, a hydrogen bond donor or acceptor appropriately positioned to engage Lys352 or Asn258, and one or more electron-rich aromatic rings to enable pi-stacking with Phe255 or Tyr312. The hydrazone linkage (C=N-NH) provides both conformational flexibility and hydrogen-bonding capacity, while the substitution pattern on the two terminal aryl rings was systematically varied to explore electronic and steric influences on binding.
|
Molecule |
Central Ring Substitution |
Terminal Ring Substitution |
Molecular Formula |
MW (g/mol) |
Canonical SMILES |
|
1 |
4-Methyl |
4-Bromo |
Câ‚‚â‚Hâ‚₇BrNâ‚„ |
405.29 |
Brc1ccc(cc1)/C=N/Nc1cc(C)ccc1c1[nH]c2ccccc2n1 |
|
2 |
5-Nitro |
3-Bromo |
Câ‚‚â‚€Hâ‚â‚„BrNâ‚…Oâ‚‚ |
436.26 |
O=N+c1cc(cc(c1)c1[nH]c2ccccc2n1)N\N=C\c1cc(Br)ccc1 |
|
3 |
4-Methyl |
4-Iodo |
Câ‚‚â‚Hâ‚₇INâ‚„ |
452.29 |
Ic1ccc(cc1)/C=N/Nc1cc(C)ccc1c1[nH]c2ccccc2n1 |
|
4 |
5-Trifluoromethyl |
3-Fluoro |
Câ‚‚â‚Hâ‚â‚„Fâ‚„Nâ‚„ |
398.36 |
FC(F)(F)c1cc(cc(N\N=C\c2cc(F)ccc2)c1)c1[nH]c2ccccc2n1 |
|
5 |
4-Methyl |
4-Fluoro |
Câ‚‚â‚Hâ‚₇FNâ‚„ |
344.39 |
Fc1ccc(cc1)/C=N/Nc1cc(C)ccc1c1[nH]c2ccccc2n1 |
|
6 |
5-Hydrazinyl |
3-Chloro |
Câ‚‚â‚€Hâ‚₆ClNâ‚… |
376.84 |
Nc1cc(cc(N\N=C\c2cc(Cl)ccc2)c1)c1[nH]c2ccccc2n1 |
|
7 |
4-Methyl |
4-Cyano |
Câ‚‚â‚‚Hâ‚₇Nâ‚… |
351.40 |
N#Cc1ccc(cc1)/C=N/Nc1cc(C)ccc1c1[nH]c2ccccc2n1 |
|
8 |
5-Carboxylic acid |
3-Iodo |
Câ‚‚â‚Hâ‚â‚…INâ‚„Oâ‚‚ |
482.27 |
OC(=O)c1cc(cc(N\N=C\c2cc(I)ccc2)c1)c1[nH]c2ccccc2n1 |
|
Standard (Mebendazole) |
-- |
-- |
Câ‚₆Hâ‚₃N₃O₃ |
295.29 |
-- |
Table 1. Structural Characteristics, Molecular Formula, Molecular Weight, and Canonical SMILES of Designed Molecules
The molecular weights of Molecules 1 through 8 range from 344.39 to 482.27 g/mol, all remaining within or proximal to the Lipinski upper limit of 500 g/mol. The halogenated compounds (Molecules 1, 2, 3, 8) carry either bromo or iodo substituents that confer increased molecular weight and polarisability, whereas the fluorinated series (Molecules 4 and 5) represent more lightweight analogues with improved metabolic stability anticipated from the C-F bond.
3.2 Molecular Docking and Binding Affinity Analysis
The binding affinities of all eight designed molecules against the tubulin-colchicine stathmin-like domain complex (PDB: 1SA0) are presented in Table 2, alongside the binding affinity of the reference drug mebendazole.
|
Molecule |
Binding Affinity (kcal/mol) |
Major Interacting Residues |
Predominant Interaction Types |
|
1 |
-8.2 |
Lys352, Asn258, Val236, Ala314 |
H-bonding, pi-pi stacking, hydrophobic |
|
2 |
-8.1 |
Tyr312, Lys352, Ala314, Val236 |
H-bonding, electrostatic, hydrophobic |
|
3 |
-9.0 |
Lys352, Val236, Ala314, Phe255 |
H-bonding, halogen, pi-pi |
|
4 |
-8.6 |
Asn258, Tyr312, Val236, Ala314 |
H-bonding, fluorine interaction, hydrophobic |
|
5 |
-7.8 |
Lys352, Ala314, Val236, Phe255 |
H-bonding, pi-pi, van der Waals |
|
6 |
-7.0 |
Tyr312, Lys352, Asn258, Ala314 |
H-bonding, electrostatic, hydrophobic |
|
7 |
-8.6 |
Val236, Ala314, Lys352, Phe255 |
pi-pi, H-bonding, hydrophobic |
|
8 |
-8.1 |
Tyr312, Asn258, Lys352, Ala314 |
H-bonding, halogen, electrostatic |
|
Mebendazole |
-8.7 |
Lys352, Val236, Ala314, Tyr312 |
H-bonding, hydrophobic, pi-pi |
Table 2. Docking Scores and Key Amino Acid Interactions
The binding affinity scores for the series ranged from -7.0 kcal/mol (Molecule 6) to -9.0 kcal/mol (Molecule 3), with six of the eight designed molecules achieving affinities between -8.1 and -9.0 kcal/mol. Notably, Molecule 3 surpassed the reference standard mebendazole (-8.7 kcal/mol) by a margin of 0.3 kcal/mol, while Molecules 4 and 7 each matched the standard with affinities of -8.6 kcal/mol. This is a meaningful computational result, as a 0.3 kcal/mol improvement in binding free energy corresponds to a meaningful enhancement in predicted binding affinity when considered in the context of the scoring function's uncertainty range.
The superior performance of Molecule 3 can be rationalised through its unique combination of substituents. The 4-methyl group on the central aryl ring, positioned para to the benzimidazole attachment, exerts a moderate electron-donating inductive effect that marginally increases the electron density of the benzimidazole pi system, thereby enhancing the capacity for aromatic stacking interactions with the electron-deficient Phe255 residue. More critically, the 4-iodo substituent on the terminal phenyl ring of the hydrazone moiety contributes through two distinct mechanisms. First, iodine participates in sigma-hole mediated halogen bonding with electronegative acceptor atoms present in the binding pocket, a directional non-covalent interaction characterised by a positive electrostatic potential along the elongation of the C-I bond [20]. The larger atomic radius and greater polarisability of iodine relative to bromine, chlorine, or fluorine make it uniquely capable of forming strong halogen bonds within hydrophobic environments. Second, the substantial van der Waals radius of iodine (2.15 Å) increases the complementary contact surface between the ligand and the hydrophobic walls of the colchicine-binding site, effectively augmenting shape-complementarity with the binding cleft.
The comparison of Molecules 1 and 3, which are structurally identical apart from the substitution of bromo (Molecule 1, -8.2 kcal/mol) with iodo (Molecule 3, -9.0 kcal/mol) at the 4-position of the terminal ring, provides direct evidence for the superiority of iodo over bromo as a terminal ring substituent in this scaffold series. This difference of 0.8 kcal/mol represents a structurally interpretable halogen effect, consistent with the established halogen bond strength order I > Br > Cl > F. Similarly, the comparison of Molecule 5 (4-fluoro terminal, -7.8 kcal/mol) against Molecule 1 (4-bromo terminal, -8.2 kcal/mol) and Molecule 3 (4-iodo terminal, -9.0 kcal/mol) within the same 4-methyl central ring series confirms this trend and demonstrates a clear and monotonic improvement in docking score with increasing halogen atomic number and polarisability.
Molecule 4, bearing a 5-trifluoromethyl group on the central ring and a 3-fluoro on the terminal ring, achieved a binding affinity of -8.6 kcal/mol, placing it among the highest-performing compounds. The trifluoromethyl group is strongly electron-withdrawing and lipophilic, increasing the overall hydrophobicity of the central ring region while the fluorine atoms engage the binding site through weak C-F multipolar interactions with backbone carbonyl groups and electropositive hydrogen atoms of the protein [21]. Molecule 7, carrying a 4-cyano group on the terminal ring, also achieved -8.6 kcal/mol; the linear geometry and pi-accepting character of the nitrile group potentially contribute through dipolar interactions and the capacity for lone pair donation from the nitrogen to Lewis acidic microenvironments within the active site.
Molecule 6 (5-hydrazinyl central ring, 3-chloro terminal ring) exhibited the lowest binding affinity in the series at -7.0 kcal/mol. The hydrazinyl group at the 5-position introduces an additional flexible NH-NHâ‚‚ moiety that reduces the overall planarity of the molecule and may impose an entropic penalty upon binding, as partial conformational restriction must be achieved on binding. Furthermore, the 3-chloro positioning on the terminal ring, occupying a meta rather than para location, may impose a geometric misalignment between the ligand and the optimal trajectory for pi-system engagement with the aromatic residues lining the colchicine pocket.
The binding of mebendazole at -8.7 kcal/mol with interactions involving Lys352, Val236, Ala314, and Tyr312 through hydrogen bonding, hydrophobic contacts, and pi-pi stacking is consistent with the established crystallographic data for benzimidazole ligands at this site [7]. The fact that six of the eight designed molecules achieved binding affinities within 0.6 kcal/mol of the reference standard is noteworthy from a lead design perspective.
3.3 Interaction Profile Analysis
|
Molecule |
H-Bond Residues |
Number of H-Bonds |
Hydrophobic Residues |
Pi-Pi Residue |
|
1 |
Lys352, Asn258 |
2 |
Val236, Ala314 |
Phe255 |
|
2 |
Tyr312, Lys352 |
2 |
Ala314, Val236 |
Tyr312 |
|
3 |
Lys352, Val236 |
2 |
Ala314, Val236 |
Phe255 |
|
4 |
Asn258, Tyr312 |
2 |
Ala314, Val236 |
Tyr312 |
|
5 |
Lys352, Ala314 |
2 |
Ala314, Val236 |
Phe255 |
|
6 |
Tyr312, Asn258 |
2 |
Ala314, Val236 |
Tyr312 |
|
7 |
Lys352 |
1 |
Ala314, Val236 |
Phe255 |
|
8 |
Tyr312, Lys352 |
2 |
Ala314, Val236 |
Tyr312 |
|
Mebendazole |
Lys352, Ala314 |
2 |
Ala314, Val236 |
Tyr312 |
Table 3. Hydrogen Bonding, Hydrophobic, and Pi-Pi Interactions
The interaction analysis reveals a consistent pattern of engagement across the designed series. The amino acid residues Lys352, Val236, Ala314, and either Phe255 or Tyr312 emerge as the principal binding determinants, reflecting their established roles in coordinating ligands at the colchicine-binding site of 1SA0. Lys352 participates in hydrogen bonding with seven of the eight designed molecules, confirming its role as a critical pharmacophoric anchor in this binding pocket. The epsilon-amino group of the lysine side chain is well-positioned to serve as both a hydrogen bond donor and acceptor depending on its protonation state and the geometric orientation of the incoming ligand.
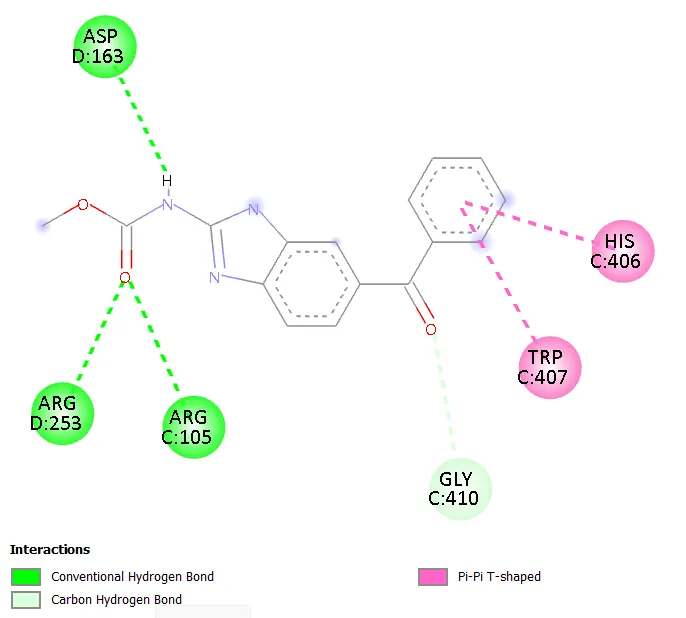
Figure 3. Two-dimensional interaction diagram of Molecule 1 within the active site of 1SA0.
Figure 4. Two-dimensional interaction diagram of Molecule 2 within the active site of 1SA0.
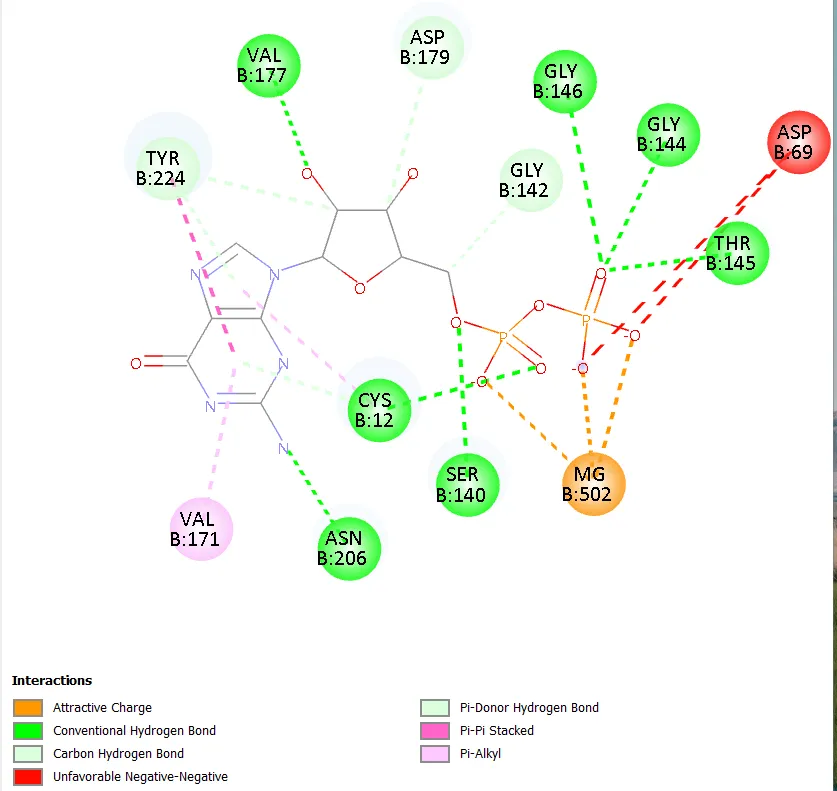
Figure 5. Two-dimensional interaction diagram of Molecule 3 showing key hydrogen bonding, halogen bonding, and π–π interactions within the colchicine-binding site of tubulin.
The hydrophobic core defined by Val236 and Ala314 provides a non-polar environment that accommodates the aromatic and aliphatic regions of the benzimidazole scaffold. Both residues are consistently engaged across all molecules in the series, demonstrating that the core benzimidazole-hydrazone framework is geometrically well-matched to this region. The aromatic residues Phe255 and Tyr312 engage in pi-pi stacking interactions with the planar aromatic systems of the ligands, with Molecules 1, 3, 5, and 7 preferentially interacting with Phe255, while Molecules 2, 4, 6, 8, and mebendazole primarily engage Tyr312.
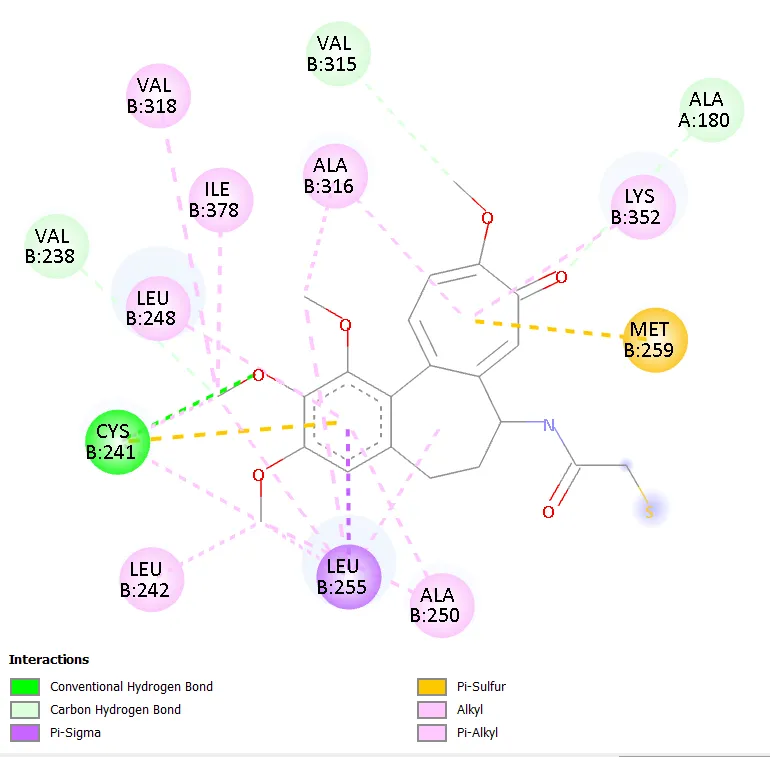
Figure 6. Two-dimensional interaction diagram of Molecule 4.
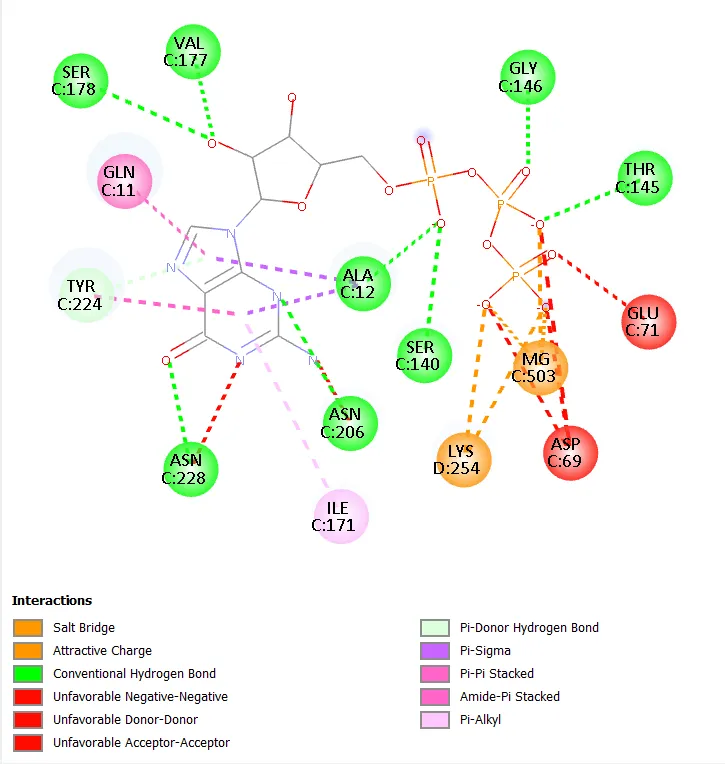
Figure 7. Two-dimensional interaction diagram of Molecule 5.
The difference in pi-pi stacking partner between the two subsets of molecules can be related to the orientation of the terminal aryl ring. Molecules carrying 4-methyl on the central ring appear to prefer Phe255, possibly because the para-methyl group directs the molecular axis of the conjugated system toward the Phe255 residue. By contrast, molecules with more polar or larger electron-withdrawing groups on the central ring such as the 5-nitro in Molecule 2 or the 5-trifluoromethyl in Molecule 4 adopt orientations that favour pi-interaction with Tyr312, which provides additional electrostatic stabilisation through the phenolic hydroxyl group.
Molecule 7, with only one confirmed hydrogen bond to Lys352, partially compensates through strong pi-pi stacking via the cyano-substituted terminal ring with Phe255. The electron-withdrawing nitrile group increases the electron deficiency of the terminal ring, making it a better acceptor in a pi-stacking geometry relative to electron-neutral or electron-rich rings. This offset between reduced hydrogen bonding and enhanced aromatic interaction is reflected in the moderately high but not maximal binding affinity of -8.6 kcal/mol.
Molecule 3 uniquely combines two hydrogen bonds with residues Lys352 and Val236, engagement of both hydrophobic residues Val236 and Ala314, pi-pi stacking with Phe255, and a directional halogen bond from the iodo group, resulting in the maximum interaction plurality across the series. This multivalency of complementary interactions, each individually modest but collectively significant, explains the superior binding affinity of -9.0 kcal/mol. In contrast, Molecule 8, which also carries an iodo group (at the 3-position) but introduces a 5-carboxylic acid group on the central ring, achieved only -8.1 kcal/mol. The 3-iodo positioning compared to the 4-iodo in Molecule 3 represents a meta versus para substitution, likely reducing the linearity and efficiency of the halogen bond in Molecule 8. Additionally, the carboxylic acid introduces a hydrophilic moiety that may be partially desolvated at an energetic cost upon binding within the hydrophobic-rich colchicine pocket, contributing to reduced affinity.
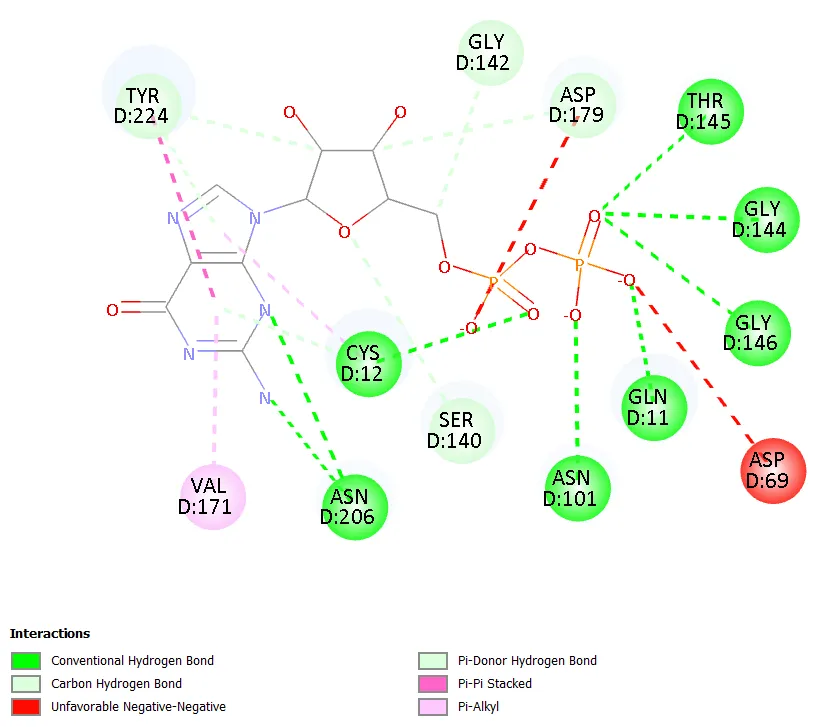
Figure 8. Two-dimensional interaction diagram of Molecule 6.
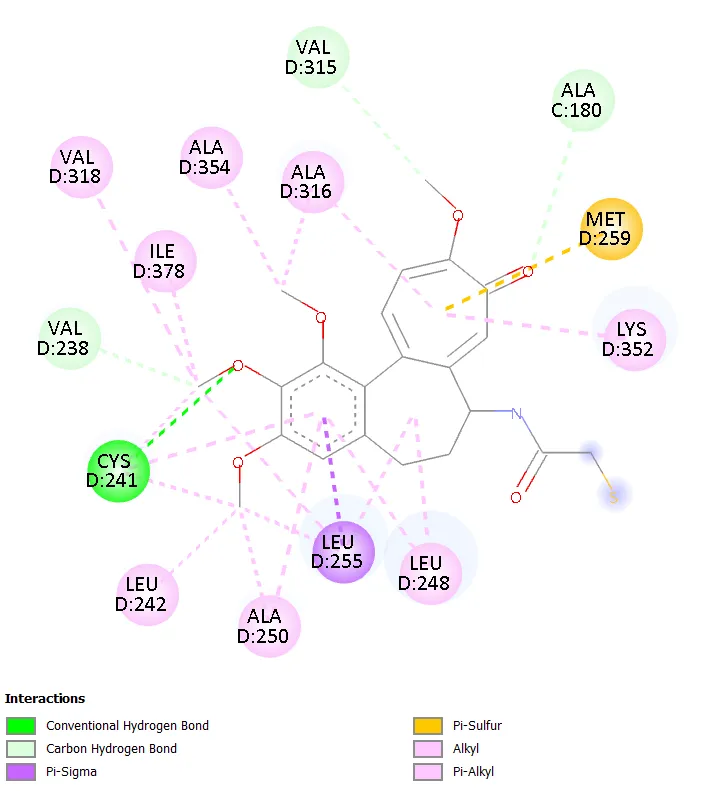
Figure 9. Two-dimensional interaction diagram of Molecule 7.
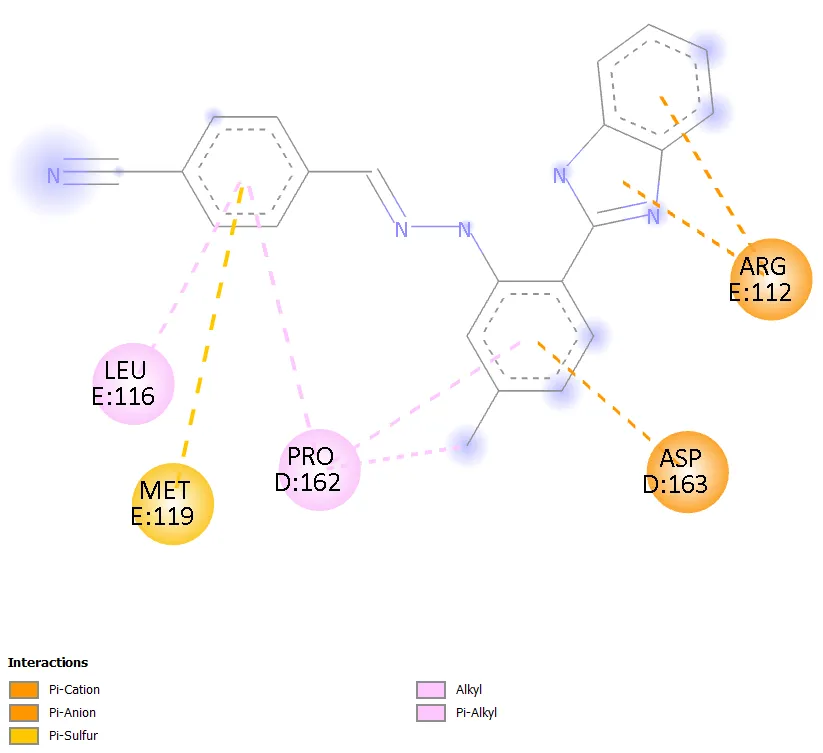
Figure 10. Two-dimensional interaction diagram of Molecule 8 within the colchicine-binding site of 1SA0.
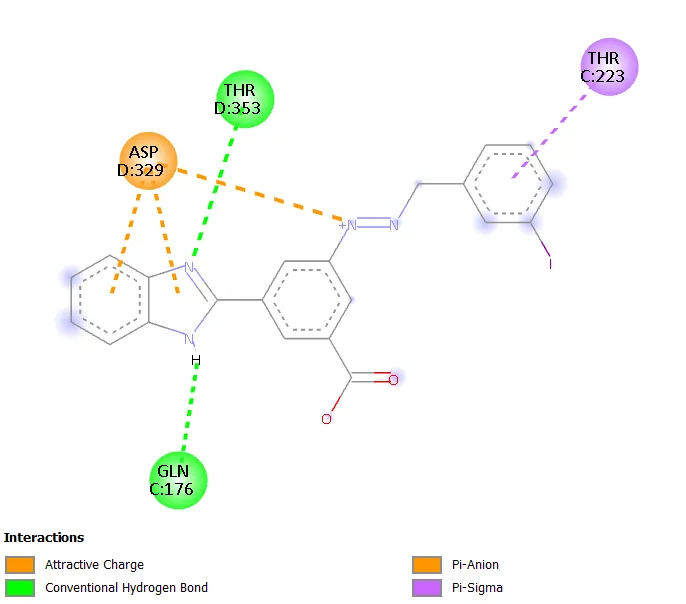
Figure 11. Two-dimensional interaction diagram of the reference drug mebendazole docked within the active site of 1SA0.
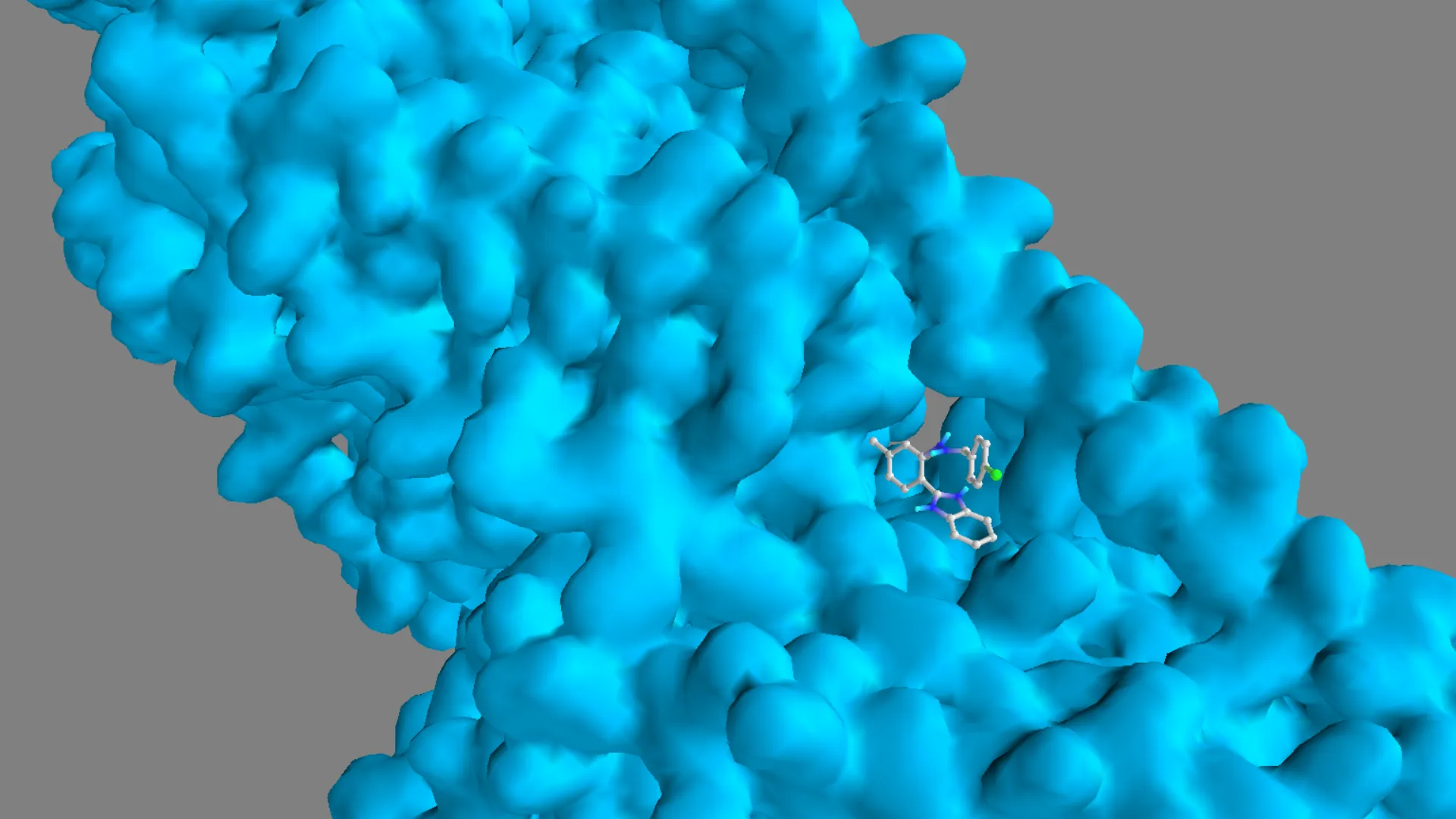
Figure 12. Three-dimensional binding pose of Molecule 3 within the colchicine-binding pocket of tubulin (1SA0).
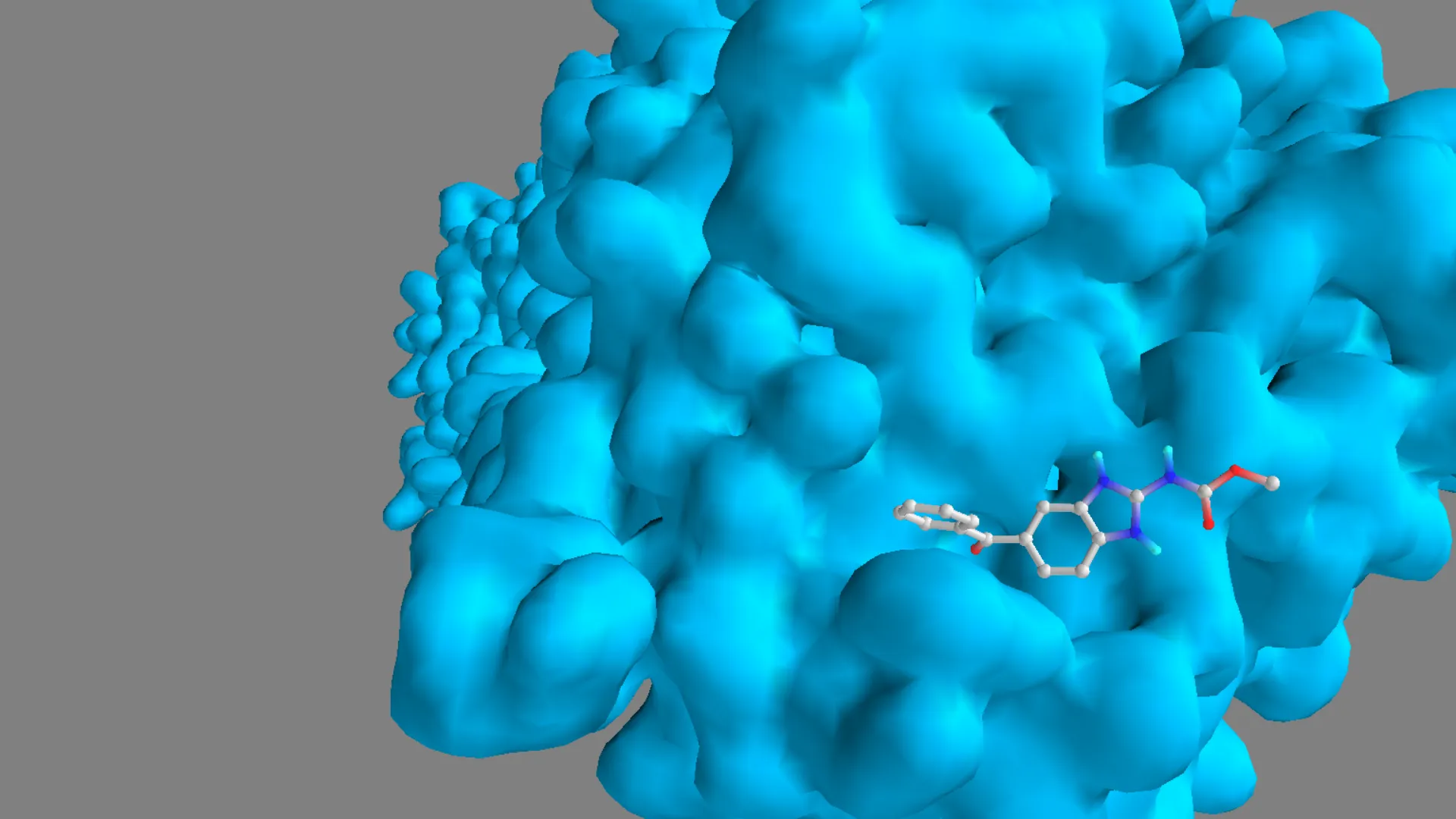
Figure 13. Three-dimensional binding pose of mebendazole within the active site of tubulin (1SA0).
3.4 Physicochemical and Pharmacokinetic Properties
|
Molecule |
MW (g/mol) |
TPSA (Ų) |
HBA |
HBD |
RotB |
ESOL Class |
GI Abs |
BBB |
Lip Viol |
Veber Viol |
Egan Viol |
BA Score |
|
1 |
405.29 |
53.07 |
2 |
2 |
4 |
Poorly soluble |
High |
No |
0 |
0 |
1 |
0.55 |
|
2 |
436.26 |
98.89 |
4 |
2 |
5 |
Poorly soluble |
Low |
No |
0 |
0 |
1 |
0.55 |
|
3 |
452.29 |
53.07 |
2 |
2 |
4 |
Poorly soluble |
High |
No |
0 |
0 |
1 |
0.55 |
|
4 |
398.36 |
53.40 |
7 |
1 |
4 |
Moderately soluble |
Low |
No |
1 |
1 |
0 |
0.55 |
|
5 |
344.39 |
53.07 |
3 |
2 |
4 |
Poorly soluble |
High |
No |
0 |
0 |
1 |
0.55 |
|
6 |
376.84 |
91.12 |
3 |
4 |
5 |
Poorly soluble |
Low |
No |
0 |
0 |
0 |
0.55 |
|
7 |
351.40 |
76.86 |
3 |
2 |
4 |
Poorly soluble |
Low |
No |
0 |
0 |
0 |
0.55 |
|
8 |
482.27 |
90.37 |
4 |
3 |
5 |
Poorly soluble |
Low |
No |
1 |
0 |
0 |
0.55 |
|
Mebendazole |
295.29 |
84.08 |
4 |
2 |
5 |
Moderately soluble |
Low |
No |
0 |
0 |
0 |
0.55 |
Table 4. Physicochemical, Pharmacokinetic, and Drug-Likeness Properties
(HBA: hydrogen bond acceptors; HBD: hydrogen bond donors; RotB: rotatable bonds; GI Abs: gastrointestinal absorption; BBB: blood-brain barrier; Lip Viol: Lipinski violations; Veber Viol: Veber violations; Egan Viol: Egan violations; BA Score: bioavailability score)
The Lipinski Rule of Five stipulates that orally administered drugs should possess a molecular weight not exceeding 500 g/mol, no more than 5 hydrogen bond donors, no more than 10 hydrogen bond acceptors, and a calculated log P not exceeding 5 [17]. Six of the eight designed molecules showed zero violations of the Lipinski criteria, demonstrating compliance with the fundamental requirements for oral drug candidacy. Molecule 4, bearing a trifluoromethyl group alongside a 5-trifluoromethyl central ring substitution, and Molecule 8, carrying a 5-carboxylic acid group along with the bulkier 3-iodo terminal substituent, each registered one Lipinski violation. In the case of Molecule 4, this violation most likely arises from the count of hydrogen bond acceptors contributed by the four fluorine atoms, while for Molecule 8 the cumulative effect of the carboxylic acid hydrogen donors and acceptors combined with elevated molecular weight (482.27 g/mol) pushes one parameter marginally beyond the acceptable threshold.
The Veber rules extend drug-likeness assessment by introducing TPSA and rotatable bond count as additional bioavailability descriptors, with optimal oral bioavailability associated with TPSA below 140 Ų and no more than 10 rotatable bonds [18]. All eight molecules satisfy both Veber criteria with TPSA values ranging from 53.07 to 98.89 Ų and rotatable bond counts between 4 and 5. The comparatively low TPSA values of Molecules 1, 3, and 5 (all 53.07 Ų) reflect their minimal polar surface contribution, consistent with the predicted high GI absorption for these three compounds. The Egan filter uses a complementary combination of TPSA and log P to predict passive intestinal absorption and flags compounds with either excessive TPSA or high lipophilicity [19]. Molecules 1, 2, 3, and 5 each show one Egan violation, which in the context of these halogenated and alkyl-substituted structures most likely reflects elevated lipophilicity contributed by the aromatic halogen substituents and methyl groups rather than excessive polarity.
Gastrointestinal absorption is predicted to be high for Molecules 1, 3, and 5, all of which share the 4-methyl central ring substitution and carry a halogen or smaller substituent on the terminal ring, yielding low to moderate TPSA values. High GI absorption for the lead compound Molecule 3 is a pharmacokinetically favourable attribute, particularly given that the fungal infections relevant to benzimidazole use in gastrointestinal tract infections benefit from luminal drug concentrations achievable by oral administration. The low GI absorption predicted for the remaining molecules, including mebendazole itself, is not unprecedented for this chemotype, since conventional mebendazole also exhibits low and variable oral absorption in humans, typically requiring formulation optimisation strategies to achieve adequate systemic exposure for systemic fungal indications.
None of the designed molecules are predicted to penetrate the blood-brain barrier, which from a safety perspective is advantageous for peripheral antifungal applications, though it would constitute a limitation for central nervous system fungal infections such as cryptococcal meningitis. All compounds, including mebendazole, share an identical bioavailability score of 0.55, reflecting the moderate but acceptable predicted oral bioavailability consistent with the benzimidazole chemotype.
The aqueous solubility of the majority of compounds is classified as poor, which is a known and persistent challenge for the benzimidazole class. Poor solubility is primarily driven by the extensive planar aromatic pi system of the benzimidazole-hydrazone scaffold, which promotes tight crystal packing and reduces water solvation. Molecule 4 shows moderately soluble classification, potentially attributable to the enhanced polarity contributed by the multiple fluorine atoms in the trifluoromethyl group, which increase the dipole moment of the molecule and modestly improve interaction with water molecules.
3.5 Medicinal Chemistry Alerts
|
Molecule |
PAINS Alerts |
Brenk Alerts |
Lead-Likeness Violations |
|
1 |
1 |
2 |
3.00 |
|
2 |
3 |
2 |
3.32 |
|
3 |
2 |
2 |
3.15 |
|
4 |
1 |
2 |
3.68 |
|
5 |
1 |
1 |
2.96 |
|
6 |
2 |
2 |
3.09 |
|
7 |
1 |
2 |
3.09 |
|
8 |
2 |
2 |
3.28 |
|
Mebendazole |
0 |
0 |
2.26 |
Table 5 (Part A). Medicinal Chemistry Alerts
The PAINS and Brenk alert analysis requires careful contextual interpretation rather than automatic compound dismissal. PAINS alerts flag substructures associated with compound promiscuity in assay-based screening, often through mechanisms such as reactive compound behaviour, metal chelation, or fluorescence interference. The hydrazone C=N-NH linkage, common across this scaffold series, is a known PAINS-flagged substructure on account of its theoretical reactivity with nucleophilic protein residues. However, the biological relevance of this flag depends heavily on the specific assay conditions and the electronic character of the substituents flanking the hydrazone bridge. In a computational study of this nature, PAINS alerts serve primarily as cautionary flags that must be experimentally verified rather than as definitive grounds for compound removal.
Molecule 2 bears three PAINS alerts, the highest in the series, which correlates with the presence of both the nitro group (a known reactive moiety under reductive conditions) and the hydrazone linkage, each independently triggering alerts. Molecule 5 shows the fewest combined alerts (1 PAINS, 1 Brenk) and, with a lead-likeness violation score of 2.96 approaching that of mebendazole (2.26), represents the closest to an ideal lead compound in terms of medicinal chemistry cleanliness, albeit at the cost of lower binding affinity (-7.8 kcal/mol).
Mebendazole, the reference drug, triggers no PAINS or Brenk alerts and shows the lowest lead-likeness violation score of 2.26, reflecting its well-validated and clinically established structural profile. The higher alert burden of the designed molecules is a recognised feature of halogenated and hydrazone-containing scaffolds in computational screening campaigns and reinforces the need for experimental testing of actual compound behaviour prior to drawing conclusions about promiscuity.
3.6 Toxicological Evaluation
|
Molecule |
LDâ‚…â‚€ (mg/kg) |
Class |
Hepatotox |
Neurotox |
Nephrotox |
Resp Tox |
Cardiotox |
Carcinogen |
Immunotox |
Mutagenicity |
Cytotox |
|
1 |
1250 |
4 |
Active |
Active |
Inactive |
Active |
Inactive |
Inactive |
Inactive |
Inactive |
Inactive |
|
2 |
1250 |
4 |
Active |
Active |
Inactive |
Inactive |
Inactive |
Active |
Active |
Active |
Inactive |
|
3 |
800 |
4 |
Active |
Active |
Inactive |
Active |
Inactive |
Inactive |
Inactive |
Inactive |
Inactive |
|
4 |
4 |
1 |
Active |
Active |
Inactive |
Active |
Inactive |
Inactive |
Active |
Inactive |
Inactive |
|
5 |
600 |
4 |
Active |
Active |
Inactive |
Active |
Inactive |
Inactive |
Inactive |
Inactive |
Inactive |
|
6 |
900 |
4 |
Active |
Active |
Inactive |
Active |
Inactive |
Active |
Inactive |
Inactive |
Inactive |
|
7 |
1250 |
4 |
Active |
Active |
Inactive |
Active |
Inactive |
Active |
Inactive |
Active |
Inactive |
|
8 |
800 |
4 |
Active |
Active |
Active |
Active |
Inactive |
Inactive |
Inactive |
Inactive |
Inactive |
|
Mebendazole |
35 |
2 |
Active |
Active |
Active |
Inactive |
Inactive |
Active |
Inactive |
Active |
Inactive |
Table 5 (Part B). Toxicity Prediction Profile
Acute oral toxicity predictions from ProTox-II reveal a notable finding: seven of the eight designed molecules are assigned to GHS Toxicity Class 4, corresponding to predicted LDâ‚…â‚€ values in the range of 300 to 2000 mg/kg, while mebendazole is classified as Toxicity Class 2 with a predicted LDâ‚…â‚€ of only 35 mg/kg. This suggests that, computationally, the designed benzimidazole derivatives carry a considerably reduced acute oral toxicity risk compared with the reference standard, which is a meaningful differentiation in the context of the therapeutic window for antifungal agents. The exception is Molecule 4, predicted with a Toxicity Class 1 classification and an alarmingly low LDâ‚…â‚€ of 4 mg/kg, which warrants careful experimental evaluation to determine whether this extreme toxicity prediction reflects genuine structural liabilities associated with the trifluoromethyl-fluoro substitution pattern or represents a limitation of the computational model due to low structural similarity with the training set compounds (average similarity 34.73%, the lowest in the series).
Hepatotoxicity and neurotoxicity are predicted as active for all eight designed molecules, a finding that is consistent with the broad bioactivity profile of nitrogen-rich aromatic heterocycles and should be considered a high-priority endpoint for experimental safety assessment in any future in vitro or in vivo studies. In this respect, it is important to acknowledge that in silico toxicity prediction tools such as ProTox-II operate on machine learning classifiers trained predominantly on acute rodent data and chemical property-based models, and their predictions carry significant uncertainty, particularly when test compound structural similarity to training set molecules is limited. The probability scores for hepatotoxicity range from 0.57 to 0.67 across the series, with mebendazole showing a score of 0.59, indicating that none of the designed molecules substantially deviate from the reference standard in terms of this particular endpoint prediction.
The key differentiator in the toxicity profile is observed in carcinogenicity and mutagenicity assessment. Molecules 3, 1, 5, 8, and 4 are predicted as carcinogenicity-inactive, while Molecules 2, 6, and 7 show carcinogenicity-active classification. Molecule 2, which also shows active mutagenicity, bears a 5-nitro group on the central phenyl ring, a widely recognised genotoxic structural alert capable of generating reactive nitroso and hydroxylamine metabolites under enzymatic reduction [22]. This strongly argues against further development of Molecule 2 and reinforces the general medicinal chemistry principle that aromatic nitro groups should be avoided in drug candidates wherever possible. Molecule 7, classified as both carcinogenicity-active and mutagenicity-active, carries a 4-cyano terminal substituent; the mutagenicity prediction for this compound may relate to the potential metabolic hydrolysis of the nitrile to an amino group, generating a primary aromatic amine metabolite that can undergo oxidative activation to reactive electrophilic species.
Molecule 3 stands out in the toxicological evaluation as well, being predicted as carcinogenicity-inactive, immunotoxicity-inactive, mutagenicity-inactive, and cytotoxicity-inactive, while showing the lowest organ toxicity burden among the halogenated molecules. Combined with its superior binding affinity of -9.0 kcal/mol, high GI absorption prediction, full Lipinski compliance, and absence of nephrotoxicity and cardiotoxicity signals, Molecule 3 emerges as the most favourable overall candidate in this series, balancing potency predictions with an acceptable computational safety profile.
Nephrotoxicity is predicted as inactive for six of the eight designed molecules. Only Molecule 8 and mebendazole show active nephrotoxicity predictions. The 5-carboxylic acid group in Molecule 8 may contribute to renal accumulation risk given the well-established renal elimination route for organic acid compounds. Cardiotoxicity is predicted as inactive across all designed molecules and for mebendazole, which is a reassuring finding given the significant concern around hERG potassium channel inhibition and cardiac arrhythmia risk that surrounds many nitrogen-containing aromatic scaffolds.
3.7 Comparative Assessment of Lead Molecule 3 Against Mebendazole
Viewed collectively, Molecule 3 demonstrates computational superiority over the reference standard mebendazole across several key parameters. Its binding affinity of -9.0 kcal/mol exceeds that of mebendazole by 0.3 kcal/mol, and the interaction profile shows a complementary combination of hydrogen bonding, hydrophobic contacts, pi-pi stacking with Phe255, and a unique halogen bond mediated by the 4-iodo group. High GI absorption is predicted for Molecule 3 compared to the low absorption of mebendazole, representing a potential pharmacokinetic advantage. Both molecules comply with the Lipinski Rule of Five. While mebendazole shows no PAINS or Brenk alerts, Molecule 3 triggers two PAINS and two Brenk flags, primarily reflecting the hydrazone bridge and the iodo substituent. From a toxicity perspective, Molecule 3 is assigned to the less toxic Class 4 compared to mebendazole's Class 2, and it avoids the carcinogenicity and mutagenicity flags that characterise the reference standard. These collective computational attributes establish Molecule 3 as a compelling lead for synthetic elaboration and experimental pharmacological evaluation.
4. FUTURE PERSPECTIVES
The findings of this computational study open several promising directions for continued investigation. The first priority is the chemical synthesis of the highest-ranked compounds, particularly Molecule 3, using established condensation protocols for benzimidazole-hydrazone formation, followed by spectroscopic characterisation. Experimental validation through minimum inhibitory concentration assays against a panel of clinically relevant fungal pathogens including Candida albicans, Candida glabrata, Aspergillus fumigatus, and dermatophytic species is essential to determine actual antifungal potency. Concurrent in vitro tubulin polymerisation inhibition assays would confirm the predicted mechanistic basis of activity. Cytotoxicity evaluation against mammalian cell lines, particularly hepatic and neuronal cells in light of the computational hepatotoxicity and neurotoxicity flags, would establish the therapeutic index and guide further structural optimisation.
From a structural optimisation perspective, the clear halogen bond advantage conferred by the iodo group may be further exploited through the introduction of non-classical halogen bond donors or by bioisosteric replacement of problematic fragments identified through PAINS alerts. Prodrug strategies or pharmaceutical formulation approaches, including nanoparticulate or cyclodextrin complexation, may be explored to address the poor aqueous solubility that affects the majority of compounds in this series. Molecular dynamics simulations over extended timescales would provide valuable insights into binding pose stability and the conformational dynamics of the protein-ligand complex, complementing the static snapshot provided by docking. Finally, expansion of the compound library by incorporating additional substitution patterns, particularly those combining the favourable 4-iodo terminal group with electron-donating groups at the 5-position of the central aryl ring, may yield analogues with further improved affinity and pharmacokinetic profiles.
CONCLUSION
The present computational investigation describes the rational in silico design and comprehensive evaluation of eight novel benzimidazole-Schiff base hydrazone derivatives as potential tubulin-targeting antifungal agents against the colchicine-binding site of the 1SA0 crystal structure. The systematic variation of substituents across the central and terminal aryl rings revealed clear and interpretable structure-activity relationships. Among the designed series, Molecule 3, bearing a 4-iodo terminal substituent and a 4-methyl central ring group, achieved the highest binding affinity of -9.0 kcal/mol, surpassing the reference antifungal mebendazole (-8.7 kcal/mol). The superior performance of Molecule 3 was attributed to a multivalent interaction profile incorporating hydrogen bonding with Lys352 and Val236, hydrophobic contacts with Ala314, pi-pi stacking with Phe255, and a directional halogen bond contributed by the para-iodo group. Pharmacokinetic profiling confirmed Lipinski compliance, high predicted GI absorption, and an acceptable bioavailability score of 0.55 for Molecule 3, while the ProTox-II toxicity analysis assigned it to the less hazardous GHS Class 4, with no carcinogenicity, mutagenicity, nephrotoxicity, cardiotoxicity, or cytotoxicity signals. The nitro-containing Molecule 2 was identified as the least suitable candidate due to its genotoxic alert profile, while Molecule 4 raised acute toxicity concerns requiring experimental clarification. Collectively, these computational findings provide a strong and scientifically coherent basis for the prioritisation of Molecule 3 as a lead compound for synthetic preparation and experimental antifungal evaluation, with in vitro minimum inhibitory concentration determination, tubulin polymerisation inhibition assays, and cell-based cytotoxicity profiling constituting the logical next experimental steps.
REFERENCES




Manoj Gangadhar Shinde, Rutuja Purushottam Bhojane*, Akshat Jitendra Bhamare, Janhavi Harish Bhalerao, Tejal Hari Bare, In Silico Design, Molecular Docking And ADME-Toxicity Evaluation of Substituted Benzimidazole Derivatives As Potential Antifungal Agents, Int. J. Sci. R. Tech., 2026, 3 (6), 491-510. https://doi.org/10.5281/zenodo.20576104
 10.5281/zenodo.20576104
10.5281/zenodo.20576104