
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Kamla Institute of Pharmaceutical Sciences, Shri Shankaracharya Professional University, Bhilai, 490020, India.
Alzheimer's disease (AD) is a progressive neurodegenerative disease with known original cholinergic dysfunction and decreased acetylcholine levels in the brain. Acetylcholinesterase (AChE) remains a validated therapeutic target for the symptomatic management of AD; however, currently available inhibitors suffer from limitations such as adverse effects and limited efficacy. Hence, alternative development of new AChE inhibitors with better pharmacological properties. In this research, a rational, thinkable series of ten novel pyrrolidine derivatives (A1-A10) was designed and confirmed by using a combined in silico approach. ChemDraw Ultra 22.2.0 and Chem3D 22.2.0 were used to design and optimize the ligands, and proprietary molecular docking programs of ArgusLab 4.0.1 were then used to dock the designed molecules with the human acetylcholinesterase (PDB ID: 4EY7). Moreover, drug-likeness, pharmacokinetic, and toxicity properties have been predicted with Swiss ADME and ProTox-II. Molecular docking results showed that the compounds A9, A3, and A2 had better docking scores as -14.3587, -13.7695, and -13.3849 kcal/mol, respectively, compared to donepezil (-13.0325 kcal/mol). Such compounds showed good interactions in the catalytic gorge of the AChE. The physicochemical properties, gastrointestinal absorption, blood – brain barrier penetration, and CNS permeability were adequate with favorable results seen during ADMET evaluation, and the toxicity was predicted to be low. In conclusion, the results indicated that pyrrolidine derivatives, notably A9, A3, and A2, appear to be good starting points for creating novel anti-Alzheimer drugs acting as inhibitors of acetylcholinesterase and should be thoroughly examined in experimental validation.
Alzheimer's disease (AD) is a progressive neurodegenerative disorder in which extracellular deposits of β-amyloid (Aβ) plaques and intracellular accumulation of hyperphosphorylated tau protein (neurofibrillary tangles) occur [1]. It is the most prevalent type of dementia in the world and has been linked to progressive cognitive deficits such as memory, language, executive functioning, and visuospatial impairments. The pathogenesis of AD is complex and involves genetic, biochemical, and environmental factors, with advancing age being the most important risk factor [2]. Alzheimer's disease (AD) is the most common neurodegenerative disease and a major contributor to dementia in older age. Pathological changes in AD can occur several years before symptoms become apparent, but the onset of clinical manifestation usually occurs after 65 years. The prevalence of AD is increasing globally due to the rapidly growing aging population. Epidemiological studies show that about 70% of dementia is caused by AD, which makes it a large public health problem and a leading cause of disability and death globally [3]. Although significant advances have been made in the understanding of the molecular mechanism of AD, useful disease-modifying therapies are restricted. Hence, new therapeutic agents with greater efficacy, safety, and pharmacokinetic properties are still of interest. This cognitive impairment that occurs in AD is attributed to a lack of acetylcholine (ACh), a neurotransmitter that is critical for learning, memory, and thinking. Acetylcholinesterase (AChE), a key enzyme present in the central nervous system, rapidly hydrolyzes acetylcholine and regulates cholinergic neurotransmission. Higher activity of AChE means lower levels of acetylcholine, which increases cognitive dysfunction. Thus, the inhibition of AChE has now been proven to be a successful approach for the symptomatic treatment of Alzheimer's disease [4].
Existing AChE inhibitors (donepezil, rivastigmine, galantamine) enhance cognitive function via increasing concentrations of AChE in the synapse [5]. But there are some drawbacks to these medicines, such as gastrointestinal side effects, hepatotoxicity, lack of effectiveness in late stages of the disease, and failure to slow the progression of the disease. These deficiencies have stimulated the search for new AChE inhibitors that are more potent, more selective, have better pharmacokinetic characteristics, and are less toxic. The heterocyclic compounds have become very significant in the medicinal field due to their wide application in various biological activities and structural versatility. Pyrrolidine derivatives constitute a privileged scaffold with a wide attention garnered due to their highly desirable pharmacological benefits and their potential binding to a variety of biological targets [6]. Pyrrolidine derivatives have been mentioned to have neuroprotective, antioxidant, anti-inflammatory, enzyme inhibitory, and central nervous system action. The properties observed indicate that pyrrolidine compounds are good potential candidates for designing novel anti-Alzheimer agents. However, the anti-Alzheimer effect of newly designed pyrrolidine derivatives, particularly that of some of them as an acetylcholinesterase inhibitor, is not yet fully explored, and thus, there exists a great scope for research. Molecular docking has become a highly promising computational method for predicting interactions of ligands with receptors, binding affinities, and molecular recognition in the active site of target molecules.
A series of new pyrrolidine derivatives was theoretically designed and assessed for anti-Alzheimer activity by targeting the acetylcholinesterase enzyme (AChE). To explore the binding ability of the designed compounds in the active site of the enzyme, molecular docking experiments were performed. Furthermore, drug-likeness and ADMET analyses were carried out to assess their pharmacokinetic behavior, oral bioavailability, and toxicity profiles. The study aims to use this integrated computational approach to find a promising pyrrolidine derivative based on their leads as a possible therapeutic molecule to be used in the treatment of Alzheimer's disease, based on which a model to be synthesized and updated in the future and tested for biological activity can be provided.
2. MATERIALS AND METHODS
2.1 Materials
The present in silico study utilized ChemDraw Ultra 22.2.0 and Chem3D 22.2.0 for ligand design and optimization, ArgusLab 4.0.1 for molecular docking analysis, SwissADME for drug-likeness and pharmacokinetic predictions, and ProTox-II for toxicity valuation of the designed pyrrolidine derivatives.
2.2 Molecular Docking Study
2.2.1 Ligand Preparation
The novel designed pyrrolidine derivatives are drawn in 2D structures, and the reference drug (Donepezil) is drawn using Chem Draw Ultra 22.2.0 software and presented in Table 1. The 2D structures were subsequently used to convert them into 3D (three-dimensional) structures using Chem3D 22.2.0. program. The structure of each ligand was optimized using the MM2 force field for energy minimization to yield a stable geometry appropriate for examination by docking studies. The optimized structures were then saved in Protein Data Bank (.pdb) format. Accurate ligand preparation was hence carried out before molecular docking analysis, paying attention to putting down proper bond order, making the correct connections between atoms, and altering molecular geometry [7-8].
|
|
||
|
Compound ID |
R |
IUPAC Name |
|
A1 |
|
(2-((4-((4-hydroxy-3-methoxybenzoyl) oxy) benzylidene) amino) ethyl) proline |
|
A2 |
|
(2-((4-(((E)-3-(3,4-dihydroxyphenyl) acryloyl) oxy) benzylidene) amino) ethyl) proline |
|
A3 |
|
(2-((4-((3-(4-hydroxy-3-methoxyphenyl) propanoyl) oxy) benzylidene) amino) ethyl) proline |
|
A4 |
|
(2-((4-((3,4,5-trihydroxybenzoyl) oxy) benzylidene) amino) ethyl) proline |
|
A5 |
|
(2-((4-((2,3,4,5,6-pentahydroxyhexanoyl) oxy) benzylidene) amino) ethyl) proline |
|
A6 |
|
(2-((4-((3-carboxy-2,3-dihydroxypropanoyl) oxy) benzylidene) amino) ethyl) proline |
|
A7 |
|
(2-((4-((4-methoxybenzoyl) oxy) benzylidene) amino) ethyl) proline |
|
A8 |
|
(2-((4-((5-carboxypentanoyl) oxy) benzylidene) amino) ethyl) proline |
|
A9 |
|
(2-((4-(cinnamoyloxy)benzylidene) amino) ethyl) proline |
|
A10 |
|
(2-((4-((3,5-dinitrobenzoyl) oxy) benzylidene) amino) ethyl) proline |
Table 1: Chemical structures of the designed pyrrolidine derivatives.
2.2.2 Protein Preparation
The three-dimensional structure of the human acetylcholinesterase (AChE) complexed with donepezil (PDB ID: 4EY7) was downloaded from the PDB webpage. The protein structure selected was the following: the residues used were 542 amino acids, and they were solved at 2.35 Å resolution. The crystallographic water molecules, cofactor molecules, metal ion molecules, and unwanted chains were deleted when preparing the structure. The protein structure was modeled by adding hydrogen atoms, side chains, and then optimized to create a suitable template to be docked with the human receptor in the next step molecular docking study [9].
2.2.3 Docking Procedure
Molecular docking studies with Argus Lab 4.0.1 version software were performed to evaluate the binding capability and molecular interaction in the active site of human acetylcholinesterase (AChE) for the designed acetylcholinesterase derivatives. The two molecules that were docked were the energy-minimized 3D models of the ligand, and the receptor was the pre-prepared and optimized 3D protein structure. The enzyme's active site is located at the center of the docking grid so as to sample the right center of the binding cavity with all molecules. The ligands were physically docked in the binding pockets of the corresponding enzymes, with several different conformations and orientations of the ligands allowed in the binding pocket in order to find the best possible docking solution for each docking. The binding energies of the complexes of the produced ligands with proteins were sorted. The binding conformation with the lowest binding energy and most favorable interaction profile was chosen for further analysis and compared to the binding conformation of the reference drug [10].
2.2.4 Molecular Visualization
Protein-ligand complexes were retrieved from Argus Lab 4.0.1, docked, and analyzed using BIOVIA Discovery Studio Visualizer. The docked poses with the highest binding energies were chosen and then input into the software for a more detailed examination of the interactions. Three-dimensional (3D) interaction illustrations were drawn to investigate the mode of binding of the ligands in the active site of the target protein [11].
2.3 In Silico ADMET and Drug-Likeness Prediction
To provide predictions of ADMET and Drug-Likeness in Silico mode. For Silico ADMET and Drug-Likeness Prediction. The design compounds were subjected to in silico calculation of ADME properties and to drug-likeness using the Swiss ADME tool from the Swiss Institute of Bioinformatics. The compounds with the best docking scores were selected for further evaluation. To assess their drug-like properties, key physicochemical parameters, including molecular weight, number of hydrogen bond donors, number of hydrogen bond acceptors, topological polar surface area, and compliance with the ‘Lipinski Rule of Five' measurement, were determined. Swiss ADME and PROTOX-II online servers were used to predict the absorption, distribution, metabolism, excretion, and toxicity (ADMET) of the selected compounds. The following computational methods were employed to make estimates of essential pharmacokinetic parameters, including gastrointestinal absorption, blood-brain barrier penetrability, and toxicity-related parameters, similar toxicity class, and LD50 [12].
3. RESULTS AND DISCUSSION
3.1 Molecular Docking Study
The molecular docking approach was used to evaluate the binding affinity and binding mode of the prepared pyrrolidine derivatives (A1-A10) in the active site of acetylcholinesterase (AChE), which is a known therapeutic target for Alzheimer's disease. The docking scores, hydrogen bond interactions, and important protein–ligand contacts were reported in Table 2. The reference drug donepezil had a docking score of -13.0325 kcal/mol, indicating that it possessed a high affinity towards the target enzyme and thus confirming the docking protocol used in the present study. The designed derivatives showed similar binding efficiency to the reference drug, with compound A9 showing the best binding score of -14.3587 kcal/mol. The higher Docking Score indicates that there are interactions in the protein-ligand binding pocket that are neither destabilizing nor disruptive to the docking process but rather stabilizing and non-disruptive. An increase in the Docking Score indicates favorable interactions in the protein ligand binding pocket, referred to as hydrogen bonding, hydrophobic, and aromatic interactions, and have conformations that do not destabilize or disrupt the docking process.
The two-dimensional interaction profile of the potent compounds complex with the reference drug is shown in Figure 3, and the three-dimensional profile of the complex in the active-site gorge is shown in Figure 4, showing good binding of the compounds to the active-site gorge. Compound A3 had the second-highest binding affinity with a docking score of -13.7695 kcal/mol and the highest number of hydrogen-bond interactions. The strong stabilization of the ligand in the binding cavity is suggested by high interaction density, potentially leading to better inhibitory potential. Likewise, for compound A2, the docking score (-13.3849 kcal/mol) was also better than that of Donepezil. The binding pattern of A2 was similar to that observed for the standard drug; this leads to a fair enzyme–ligand complementarity and efficient recognition of the active site. The rest of the derivatives had docking scores from -11.2881 to -12.1660 kcal/mol, which suggests having moderate to good binding affinity towards AChE. Among those compounds, compound A1 had the highest docking score of -12.1660kcal/mol, and compounds A4, A8, and A10 had an average number of hydrogen-bond interactions, which indicates a favorable binding stability. These compounds exhibited lower binding energies than A2, A3, and A9, but the nature of their interactions suggested that the pyrrolidine motif has the potential to form favorable contacts in the active site of the enzyme. Comparative analysis showed that comparable binding regions to those of the reference drug were found for most of the derivatives, and that residues within the catalytic and peripheral binding domains of AChE were observed for them. These interactions are thought to be important for good enzyme inhibition and have substantial effects on the stability of the enzyme-ligand complexes. Determination of the binding orientations further confirms the efficiency of efficient binding of the designed molecules within the gorge of the active site, thus preventing substrate binding to the active site and potentially inhibiting its activity.
Overall, as described in Table 2, the designed pyrrolidine derivatives illustrate promising inhibitory potential against the enzyme acetylcholinesterase. For all compounds, A9, A3, and A2 turned out to be the most promising and had higher binding affinity than the standard compound, donepezil. Results revealed that the pyrrolidine motif is considered a promising skeleton for the generation of new anti-Alzheimer drugs.
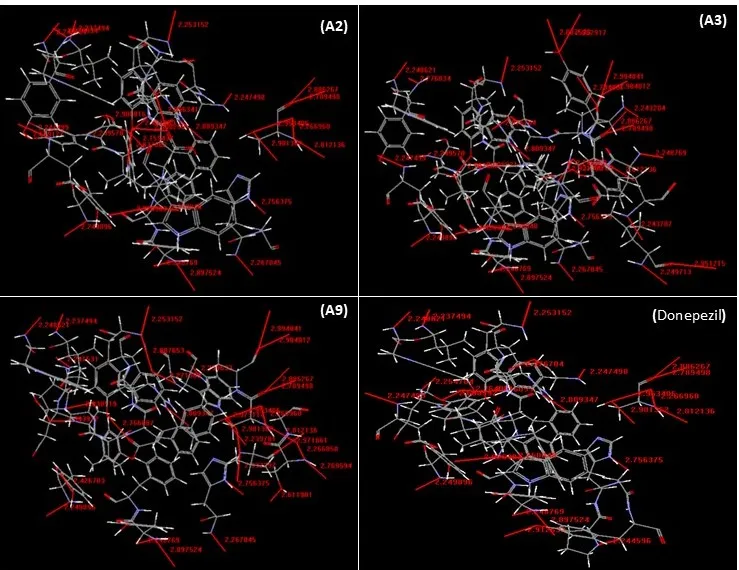
Figure 3. 2D interaction diagrams showing the binding interactions of the selected pyrrolidine derivatives (A2, A3, and A9) and the standard drug donepezil with acetylcholinesterase (AChE, PDB ID: 4EY7). The figures highlight the major intermolecular contacts involved in stabilizing the protein–ligand complexes within the enzyme active site.
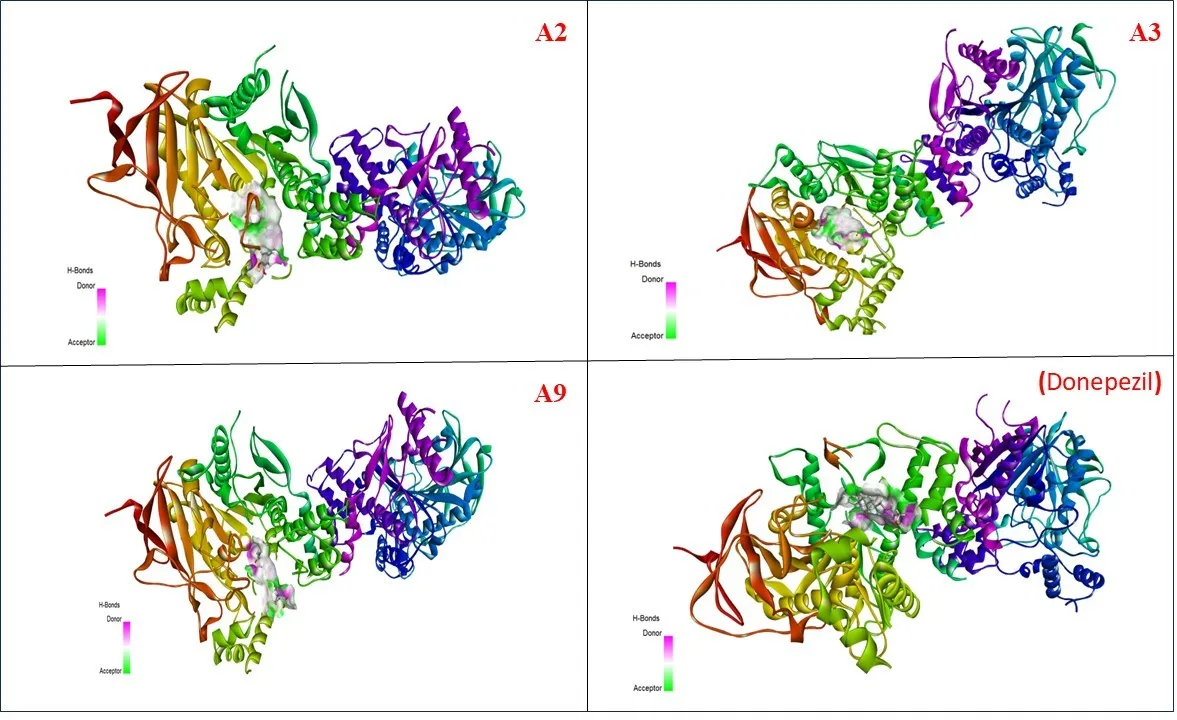
Figure 4. Three-dimensional binding poses of potent pyrrolidine derivatives (A2, A3, and A9) and donepezil in the active site of acetylcholinesterase (PDB ID: 4EY7).
|
Compound ID |
Docking Score (Kcal/mol) |
No. of H - bonds |
Interaction of Amino Acids |
|
Donepezil |
-13.0325 |
20 |
296ARG, 74ASP, 292GLU, 121GLY, 447HIS, 289LEU, 295PHE, 297PHE, 338PHE, 203SER, 293SER, 86TRP, 286TRP, 124TYR, 337TYR, 341TYR, 294VAL |
|
A1 |
-12.166 |
20 |
296ARG, 292GLU, 121GLY, 447HIS, 289LEU, 295PHE, 297PHE, 338PHE, 203SER, 293SER, 286TRP, 124TYR, 341TYR, 294VAL |
|
A2 |
-13.3849 |
20 |
296ARG, 121GLY, 447HIS, 289LEU, 295PHE, 297PHE, 338PHE, 203SER, 293SER, 86TRP, 286TRP, 124TYR, 337TYR, 341TYR, 294VAL |
|
A3 |
-13.7695 |
25 |
296ARG, 74ASP, 121GLY, 447HIS, 295PHE, 297PHE, 338PHE, 203SER, 286SER, 124TYR, 337TYR, 341TYR, 294VAL |
|
A4 |
-11.9118 |
21 |
296ARG, 74ASP, 292GLU, 121GLY, 447HIS, 289LEU, 295PHE, 297PHE, 338PHE, 203SER, 293SER, 86TRP, 286TRP, 124TYR, 337TYR, 341TYR, 294VAL |
|
A5 |
-11.424 |
20 |
296ARG, 74ASP, 121GLY, 447HIS, 289LEU, 295PHE, 297PHE, 338PHE, 203SER, 293SER, 86TRP, 286TRP, 124TYR, 337TYR, 341TYR, 294VAL |
|
A6 |
-11.2881 |
19 |
296ARG, 74ASP, 121GLY, 447HIS, 289LEU, 295PHE, 297PHE, 338PHE, 203SER, 293SER, 86TRP, 286TRP, 124TYR, 341TYR, 294VAL |
|
A7 |
-11.2906 |
19 |
296ARG, 74ASP, 121ASP, 447HIS, 289LEU, 295PHE, 297PHE, 338PHE, 203SER, 293SER, 86TRP, 286TRP, 124TYR, 337TYR, 341TYR, 294VAL |
|
A8 |
-11.8548 |
21 |
296ARG, 292GLU, 121GLY, 447HIS, 289LEU, 295PHE, 297PHE, 338PHE, 203SER, 86TRP, 286TRP, 124TYR, 337TYR, 341TYR, 294VAL |
|
A9 |
-14.3587 |
20 |
296ARG, 447HIS, 289LEU, 295PHE, 297PHE, 338PHE, 203SER, 293SER, 286TRP, 124TYR, 337TYR, 341TYR, 294VAL |
|
A10 |
-12.0461 |
23 |
296ARG, 292GLU, 121GLY, 447HIS, 289LEU, 295PHE, 297PHE, 338PHE, 203SER, 86TRP, 124TYR, 337TYR, 341TYR, 294VAL |
Table 2. Molecular docking parameters and binding interactions of the designed pyrrolidine derivatives and donepezil with acetylcholinesterase.
3.2 In Silico ADMET and Drug-Likeness Prediction
In silico ADMET prediction tools were used to determine the physicochemical properties and pharmacokinetics of the designed pyrrolidine derivatives for their potential use as anti-Alzheimer drug candidates. The physicochemical parameters calculated, such as Molecular weight (MW), hydrogen-bond donors/acceptors, rotatable bonds, bioavailability, synthetic accessibility score, drug-likeness score, and predicted toxicity values, are tabulated in Table 3, and the pharmacokinetic parameters are summarized in Table 4. The designed weights of various drug molecules ranged from 390.43 to 456.41 g/mol and were within the acceptable molecular weight range as per the rule of Lipinski for orally active drug candidates. Molecular hydrophilicity parameters such as hydrogen-bond acceptors and donors were within acceptable limits for most compounds; these are important for molecular recognition and interaction with biological targets. Moreover, the moderate level of rotatable bonds of these derivatives indicates that there is a reasonable balance between the flexibility of the molecule and the inherent stability, this being favorable to bind to the receptor and permeate the membranes. All the compounds scored positively in drug-likeness evaluation, suggesting some of them might have potential to become drug candidates. Of all the series, the compound A1 had the highest level of drug-likeness (1.33) and was followed by A4 (1.28) and A2 (1.06), demonstrating more attractive structural properties in comparison to drug development. The oral absorption probability of most derivatives was about 0.55, which is considered a reasonable absorption likelihood.
In contrast, compounds A5 and A6 had low bioavailability values, suggesting reduced systemic circulation when given orally. The range of the calculated synthetic accessibility values was between 3.47 and 4.66, which means it is of moderate synthetic feasibility. The synthetic accessibility scores of the compounds A7, A8, and A4 were relatively low, indicating that these derivatives might be more readily synthesized than those in the series. These properties are beneficial for optimization and further experimental activities. The degree of toxicity was found to be highly variable for the compounds when toxicity was assessed using estimates of LD50. Among the 3 compounds, compound A3 had the lowest acute toxicity prediction with the highest reported LD50 value (5300 mg/kg). Similarly, compounds A9 (2900 mg/kg), A2 (1500 mg/kg), and A4 (1500 mg/kg) displayed relatively low toxicity. The results indicated that compound A8 was comparatively more toxic, with the lowest LD50 (230 mg/kg), which indicated a narrow safety margin. The pharmacokinetic study shown in Table 4 exhibited that the majority of compounds had high gastrointestinal tract (GIT) absorption, which is the first requirement for drug absorption in oral administration. A1, A2, A3, A4, A7, A8, and A9 were predicted to have high intestinal absorption, while A5, A6, and A10 were predicted to have low or weak intestinal absorption. Both the high GIT absorption achieved by the majority of the derivatives confirms their potential oral bioavailability. Permeability of therapeutic agents across the blood-brain barrier (BBB) is one of the most important factors that must be met for drug molecules to gain entry to the central brain. All of the designed pyrrolidine derivatives had positive BBB permeability values, suggesting that they can permeate the BBB. Values of BBB penetration were particularly high for the compounds A5, A6, A9, and A10, indicating high distribution to brain tissues. Similarly, all compounds displayed a favorable CNS permeability score indicative of neuronal access, relevant to Alzheimer's diseases and other Central Nervous System (CNS) related applications. The clearance predicted differed between the compounds (ranging from 0.17 to 0.56 log mL/min/kg); A3 had the highest value, and A10 had the lowest. The results suggest inter-individual variability in metabolic and excretory characteristics, which can affect drug exposure and duration of action. However, the measured elimination profiles were preserved and remained within tolerable ranges of those for therapeutic candidates.
Overall, ADMET and drug-likeness show that most of the pyrrolidine derivatives designed have good physicochemical and pharmacokinetic properties. Particularly, compounds A2, A3, and A9 possessed an excellent combination of drug-likeness, oral absorption ability, blood-brain barrier permeability, CNS penetration, and safety profile. These molecular docking findings align with the results presented in this study, with some of the same molecules exhibiting high binding affinity for acetylcholinesterase. Therefore, A2, A3, and A9 can be considered the most promising lead candidates for further optimization and experimental validation as potential anti-Alzheimer agents.
|
Compound ID |
MW (g/mol) |
nHA |
nAHA |
nRB |
nHBA |
nHBD |
BA |
SA |
Drug likeness |
|
A1 |
412.44 |
30 |
12 |
9 |
8 |
2 |
0.55 |
3.67 |
1.33 |
|
A2 |
424.45 |
31 |
12 |
9 |
8 |
3 |
0.55 |
3.8 |
1.06 |
|
A3 |
440.49 |
32 |
12 |
11 |
8 |
2 |
0.55 |
3.79 |
0.77 |
|
A4 |
414.41 |
30 |
12 |
8 |
9 |
4 |
0.55 |
3.55 |
1.28 |
|
A5 |
440.44 |
31 |
6 |
12 |
11 |
6 |
0.17 |
4.66 |
0.34 |
|
A6 |
394.38 |
28 |
6 |
10 |
10 |
4 |
0.11 |
4 |
0.44 |
|
A7 |
396.44 |
29 |
12 |
9 |
7 |
1 |
0.55 |
3.47 |
0.86 |
|
A8 |
390.43 |
28 |
6 |
12 |
8 |
2 |
0.56 |
3.51 |
0.40 |
|
A9 |
392.45 |
29 |
12 |
9 |
6 |
1 |
0.55 |
3.66 |
0.61 |
|
A10 |
456.41 |
33 |
12 |
10 |
10 |
1 |
0.55 |
3.78 |
0.53 |
Table 3. Physicochemical profile and drug-likeness properties of the designed pyrrolidine derivatives.
MW: Molecular weight, nHA: No. of H-bond, nAHA: No. of Aromatic H-bond, nRB: No. of Rotable bond, nHBA: No. of H-bond Acceptor, nHBD: No. of H-bond donor, BA: Bioavailability, SA: Synthetic Accessibility, LD50: Lethal dose 50.
|
Compound ID |
GIT Absorption (High /Low) |
Distribution |
Elimination |
Toxicity |
|
|
BBB Numeric (log BB) |
CNS Numeric (log PS) |
Numeric (log ml/min/kg) |
LD50 (mg/kg) |
||
|
A1 |
High |
1.377 |
3.371 |
0.43 |
650 |
|
A2 |
High |
1.435 |
3.409 |
0.36 |
1500 |
|
A3 |
High |
1.302 |
3.377 |
0.56 |
5300 |
|
A4 |
High |
1.825 |
3.783 |
0.47 |
1500 |
|
A5 |
Low |
2.392 |
4.991 |
0.52 |
800 |
|
A6 |
Low |
1.877 |
4.365 |
0.33 |
800 |
|
A7 |
High |
1.038 |
2.832 |
0.43 |
800 |
|
A8 |
High |
1.458 |
3.539 |
0.26 |
230 |
|
A9 |
High |
1.777 |
2.612 |
0.38 |
2900 |
|
A10 |
Low |
1.837 |
3.147 |
0.17 |
1000 |
Table 4. Pharmacokinetic and toxicity parameters of the designed pyrrolidine derivatives.
GIT: Gastrointestinal tract, BBB: Blood-brain barrier, CNS: Central nervous system.
CONCLUSION
In the present study, a series of novel pyrrolidine derivatives was successfully designed and computationally evaluated as potential anti-Alzheimer agents targeting acetylcholinesterase (AChE). Molecular docking studies showed good binding interactions with the designed molecules in the active site of AChE, and compounds A9, A3, and A2 showed higher binding affinity than the reference drug donepezil. The observed docking results indicate that this pyrrolidine skeleton has good prospects to develop effective AChE inhibitors. In addition, the in silico ADMET and drug-likeness predictions suggest that most of the generated derivatives are suitable in terms of their physicochemical properties, good oral bioavailability, blood-brain barrier penetration potential, CNS penetration, and safety features. Based on the balance of binding affinity, pharmacokinetic properties, and predicted toxicity profile evaluated, A2, A3, and A9 were identified as the most promising lead candidates. Overall, the findings of this study highlight the potential of pyrrolidine-based derivatives as promising therapeutic candidates for Alzheimer's disease. The results, however, are based on predictions of computer models and should be validated experimentally. Further work will be directed towards the chemical preparation of the most interesting modified compounds, and before their structural characterization by spectroscopic studies. The synthesized compounds will then undergo in vitro biological activity evaluation, such as acetylcholinesterase inhibitory activity and in vitro neuroprotective studies, to verify the anti-Alzheimer activity. Currently, in vivo pharmacological and toxicological studies can be in line with the development of these pyrrolidine derivatives as effective therapeutic agents for the treatment of Alzheimer's disease.
REFERENCES

Mousmi Sahu, Anju Daharia*, In Silico Exploration of Novel Pyrrolidine Derivatives Targeting Alzheimer's Disease Through Molecular Docking and Pharmacokinetic Evaluation, Int. J. Sci. R. Tech., 2026, 3 (6), 1102-1111. https://doi.org/10.5281/zenodo.20757289
 10.5281/zenodo.20757289
10.5281/zenodo.20757289