
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
K. V. N. Naik S. P. Sanstha’s, Institute of Pharmaceutical Education & Research, Nashik, 422002, Maharashtra, India.
The emergence of antimicrobial resistance has created an urgent need for the development of new antibacterial agents with improved efficacy and safety. DNA gyrase is an essential bacterial enzyme involved in DNA replication and transcription and represents an important target for antibacterial drug discovery. In the present study, six novel quinolone derivatives (QD-1 to QD-6) were designed and computationally evaluated as potential antibacterial agents against Mycobacterium tuberculosis DNA gyrase (PDB ID: 3IFZ) using molecular docking, ADMET prediction, and toxicity analysis. The chemical structures of the designed compounds were prepared using ChemSketch and molecular docking was performed using PyRx integrated with AutoDock Vina. Protein-ligand interactions were analysed using BIOVIA Discovery Studio Visualizer. Pharmacokinetic and drug-likeness properties were predicted using SwissADME, while toxicity profiles were evaluated using ProTox-II. The docking scores of the designed compounds ranged from -6.7 to -7.4 kcal/mol. Among all compounds, QD-5 demonstrated the highest binding affinity with a docking score of -7.4 kcal/mol, which was slightly better than the standard drug Ciprofloxacin (-7.3 kcal/mol). Interaction analysis revealed that QD-5 formed stable conventional hydrogen bonds with ASN A:279 and ARG A:98 along with hydrophobic and halogen interactions within the active site of DNA gyrase. ADMET analysis showed that all compounds satisfied Lipinski’s Rule of Five and exhibited high gastrointestinal absorption. Toxicity prediction indicated acceptable toxicity profiles with no predicted hepatotoxicity or cardiotoxicity for the designed compounds. The overall findings suggest that QD-5 may serve as a promising lead compound for the future development of antibacterial agents targeting DNA gyrase. However, further in vitro and in vivo studies are required to validate these computational results.
Bacterial infections remain among the leading causes of morbidity and mortality worldwide. According to the World Health Organization (WHO), antimicrobial resistance (AMR) represents one of the most pressing threats to global public health. Among bacterial pathogens of concern, Mycobacterium tuberculosis (M. tuberculosis) is particularly significant, being responsible for tuberculosis (TB), a disease that continues to claim over a million lives annually. The emergence of multidrug-resistant tuberculosis (MDR-TB) and extensively drug-resistant tuberculosis (XDR-TB) has substantially reduced the effectiveness of available therapeutic regimens, making the discovery of new antibacterial agents a research priority of urgent importance [1].
Quinolone antibacterials constitute one of the most widely used and clinically successful classes of synthetic antimicrobial agents. Since the discovery of nalidixic acid in the 1960s, the quinolone scaffold has undergone extensive structural modification to produce successive generations of compounds with broader antibacterial spectra, improved pharmacokinetic profiles, and enhanced potency [2,3]. Fluoroquinolones such as ciprofloxacin, levofloxacin, and moxifloxacin are currently used in clinical practice for a wide range of infections, including respiratory tract infections, urinary tract infections, and, in some cases, TB. However, increasing resistance to fluoroquinolones, particularly in M. tuberculosis strains, has highlighted the need for new quinolone derivatives with improved activity and overcome existing resistance mechanisms [4].
The primary molecular targets of quinolone antibacterials are bacterial type II topoisomerases, specifically DNA gyrase and topoisomerase IV. DNA gyrase is an enzyme that belongs to the topoisomerase family and plays an indispensable role in bacterial DNA replication, transcription, and cell division. It introduces negative supercoils into DNA, thereby relieving torsional stress generated ahead of the replication fork. Unlike mammalian cells, which lack a functional equivalent, bacterial cells depend on DNA gyrase for survival, making it an attractive and selective antibacterial drug target [5]. In M. tuberculosis, DNA gyrase is uniquely important because this organism lacks topoisomerase IV, meaning that gyrase carries out both the DNA relaxation and supercoiling functions essential for bacterial viability [6].
In silico computational drug design refers to the use of computer-based methods to identify, design, and evaluate potential drug candidates before they are synthesized and tested in the laboratory. Among these methods, molecular docking is a widely used computational technique that predicts the preferred binding orientation and affinity of a small molecule (ligand) within the active site of a target protein. It provides important information about binding energy, intermolecular interactions, and the spatial arrangement of the ligand-protein complex. Molecular docking has proven to be a cost-effective and time-efficient strategy for screening large numbers of compounds and prioritising candidates for experimental evaluation [7].
Alongside molecular docking, the prediction of absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties has become an integral component of the modern drug discovery workflow. ADMET prediction tools such as SwissADME allow researchers to evaluate the drug-likeness of candidate compounds based on established pharmacokinetic parameters and rules such as Lipinski's Rule of Five [8]. In parallel, toxicity prediction platforms such as ProTox-II provide computational estimates of acute oral toxicity and organ-specific toxic endpoints, helping to identify potentially harmful candidates early in the discovery process [9, 10].
The present study reports the design of six novel quinolone derivatives based on structural modifications of the core quinolone scaffold, followed by their computational evaluation against DNA gyrase of M. tuberculosis (PDB ID: 3IFZ) through molecular docking, ADMET profiling, and toxicity prediction. The study aims to identify the most promising derivative that could serve as a lead molecule for future antibacterial drug development.
2. AIM AND OBJECTIVES
Aim
The primary aim of this study was to design and evaluate novel quinolone derivatives as potential antibacterial agents against M. tuberculosis through computational drug design approaches.
Objectives
3. MATERIALS AND METHODS
3.1 Target Protein Selection and Preparation
The three-dimensional crystal structure of DNA gyrase from M. tuberculosis was retrieved from the RCSB Protein Data Bank (https://www.rcsb.org) using the accession code PDB ID: 3IFZ [11]. This structure represents the breakage-and-reunion domain of the gyrase reaction core, solved at 2.7 angstrom resolution. The downloaded protein structure was opened in PyRx (version 0.8) and subjected to preparation steps that included the removal of water molecules, addition of polar hydrogen atoms, assignment of Gasteiger charges, and energy minimisation. The prepared macromolecule was saved in the PDBQT format, which is required by AutoDock Vina for docking calculations.
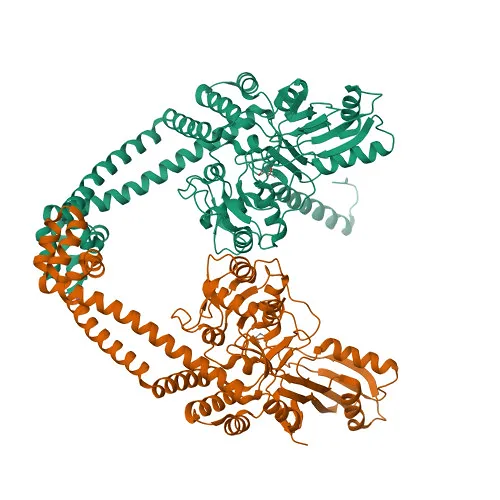
Figure 1. Three-dimensional crystal structure of Mycobacterium tuberculosis DNA gyrase (PDB ID: 3IFZ).
3.2 Design and Preparation of Quinolone Derivatives
Six quinolone derivatives were designed by modifying the core 4-oxo-1,4-dihydroquinoline-3-carboxylic acid scaffold, which is the fundamental pharmacophoric unit responsible for antibacterial activity in the quinolone class. Structural diversity was introduced by varying substituents at positions 1, 6, and 7 of the quinolone rings. Modifications included introduction of alkyl groups (methyl, ethyl, cyclopropyl) at position 1 (N-1), halogen substituents (fluorine, chlorine) or methoxy groups at position 6 (C-6), and different nitrogen-containing heterocyclic groups (morpholine, piperazine, thiomorpholine) at position 7 (C-7). These substitution patterns are well established as important determinants of antibacterial potency and pharmacokinetic behaviour in fluoroquinolones [2,3].
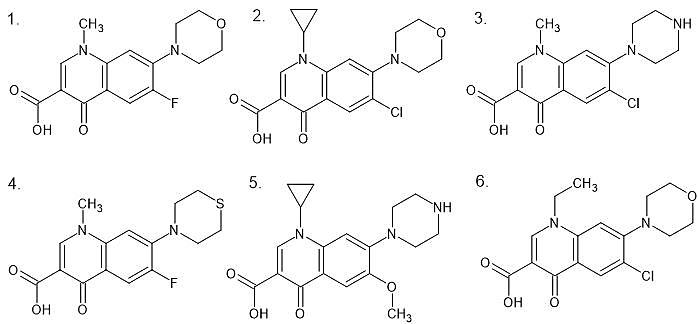
Figure 2. Chemical structures of designed quinolone derivatives (QD-1 to QD-6).
The two-dimensional structures of the designed compounds and the reference drug ciprofloxacin were drawn using ChemSketch Professional. The chemical structures were exported in SDF or MOL format and then imported into PyRx for three-dimensional structure generation. Energy minimisation was performed on each ligand using the universal force field (UFF) within the Open Babel module integrated in PyRx. All ligands were subsequently converted to PDBQT format for docking calculations.
3.3 Molecular Docking Using PyRx and AutoDock Vina
Molecular docking was carried out using AutoDock Vina (version 1.1.2) as implemented within the PyRx graphical interface [7,12]. The docking grid box was defined to encompass the active site of DNA gyrase. Grid box dimensions and coordinates were set to cover the binding pocket of the protein based on the location of the co-crystallised ligand or the catalytic residues known in the literature. The grid box was set with dimensions and centre coordinates sufficient to accommodate all ligands within the active site region. An exhaustiveness value of 8 was used, which is the standard setting for AutoDock Vina and provides a reasonable balance between computational speed and accuracy. Docking was performed for all six designed compounds and for ciprofloxacin. The binding energy (docking score, expressed in kcal/mol) of the lowest energy conformation was recorded for each compound. Lower (more negative) docking scores indicate stronger predicted binding affinity.
3.4 Protein-Ligand Interaction Analysis
After docking, the binding poses of the compounds within the DNA gyrase active site were exported and visualised using BIOVIA Discovery Studio Visualizer (Dassault Systems, version 21.1.0) [16]. Interaction diagrams were generated to identify and characterise the types of intermolecular contacts between each ligand and the receptor residues. These interactions included conventional hydrogen bonds, carbon-hydrogen bonds, hydrophobic interactions, pi-pi stacking, pi-alkyl interactions, halogen interactions, and any unfavourable contacts. The identity of the interacting amino acid residues was recorded for each compound.
3.5 ADMET and Drug-Likeness Prediction Using SwissADME
The absorption, distribution, metabolism, excretion, and toxicity-related properties of the designed compounds were predicted using the SwissADME web server (http://www.swissadme.ch), developed by the Molecular Modelling Group at the Swiss Institute of Bioinformatics [8]. For each compound, the SMILES notation was input into the server. The following parameters were evaluated: molecular weight (MW), topological polar surface area (TPSA), number of hydrogen bond acceptors (HBA), number of hydrogen bond donors (HBD), number of rotatable bonds (RB), consensus Log P (lipophilicity), gastrointestinal (GI) absorption, blood-brain barrier (BBB) permeability, adherence to Lipinski's Rule of Five, and the bioavailability score. Lipinski's Rule of Five compliance was considered to indicate oral drug-likeness [10].
3.6 Toxicity Prediction Using ProTox-II
In silico toxicity prediction was performed for all designed compounds and ciprofloxacin using ProTox-II (https://tox-new.charite.de/protox_II]), a web-based tool developed at Charite Universitatsmedizin Berlin [9]. SMILES strings of each compound were submitted to the server. The predicted acute oral toxicity expressed as LD50 (mg/kg) and GHS toxicity class (I to VI) were recorded. ProTox-II also provides predictions for organ-specific endpoints including hepatotoxicity, neurotoxicity, nephrotoxicity, respiratory toxicity, cardiotoxicity, carcinogenicity, immunotoxicity, mutagenicity, and cytotoxicity.
4. RESULTS
4.1 Target Protein: DNA Gyrase (PDB ID: 3IFZ)
The three-dimensional crystal structure of DNA gyrase from M. tuberculosis (PDB ID: 3IFZ) was successfully retrieved from the RCSB Protein Data Bank. This structure represents the breakage-and-reunion domain of the gyrase reaction core and contains two protein chains (chain A and chain B), resolved at 2.7 angstrom resolution. DNA gyrase consists of two subunits, GyrA and GyrB. The GyrA subunit is responsible for DNA cleavage and religation, while GyrB contains the ATPase domain. The active site is located at the interface of the two subunits and includes key catalytic residues that are conserved across bacterial species, making it an appropriate target for antibacterial drug design.
4.2 Designed Quinolone Derivatives and Docking Scores
Six novel quinolone derivatives designated QD-1 to QD-6 were designed and their molecular docking scores against DNA gyrase are presented in Table 1. The docking scores of the designed compounds ranged from -6.7 kcal/mol (QD-3) to -7.4 kcal/mol (QD-5). The standard drug ciprofloxacin produced a docking score of -7.3 kcal/mol. QD-5 showed the best binding affinity among all the studied compounds, with a docking score of -7.4 kcal/mol, which was marginally better than that of ciprofloxacin. QD-2 matched the docking score of ciprofloxacin at -7.3 kcal/mol. QD-1 (-7.1 kcal/mol) and QD-4 (-7.2 kcal/mol) also showed reasonably strong binding. QD-6 (-6.8 kcal/mol) and QD-3 (-6.7 kcal/mol) had the comparatively lower binding affinities among the series. The structural and docking details of all compounds are summarised in Table 1.
|
Compound |
Chemical Name |
Molecular Formula |
Molecular Weight (g/mol) |
Docking Score (kcal/mol) |
|
QD-1 |
6-fluoro-1-methyl-7-(morpholin-4-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid |
Câ‚â‚…Hâ‚₆FN₃O₃ |
305.31 |
-7.1 |
|
QD-2 |
6-chloro-1-cyclopropyl-7-(morpholin-4-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid |
Câ‚₇Hâ‚₇ClNâ‚‚Oâ‚„ |
332.33 |
-7.3 |
|
QD-3 |
6-chloro-1-methyl-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid |
Câ‚₇Hâ‚‚â‚€FN₃O₃ |
333.36 |
-6.7 |
|
QD-4 |
6-fluoro-1-methyl-4-oxo-7-(thiomorpholin-4-yl)-1,4-dihydroquinoline-3-carboxylic acid |
Câ‚₇Hâ‚₇FNâ‚‚O₃S |
349.34 |
-7.2 |
|
QD-5 |
1-cyclopropyl-6-methoxy-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid |
Câ‚₇Hâ‚‚â‚€N₃Oâ‚„ |
363.80 |
-7.4 |
|
QD-6 |
6-chloro-1-ethyl-7-(morpholin-4-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid |
Câ‚₇Hâ‚₆FN₃Oâ‚…S |
393.39 |
-6.8 |
|
Ciprofloxacin |
Standard Drug |
Câ‚₇Hâ‚₈FN₃O₃ |
331.35 |
-7.3 |
Table 1. Chemical details and molecular docking scores of designed quinolone derivatives against DNA gyrase.
* Molecular formulas are as provided in structural data. MW = Molecular Weight. Docking score unit: kcal/mol. Ciprofloxacin used as standard reference drug.
4.3 Protein-Ligand Interaction Analysis
The binding interactions between each designed compound and the DNA gyrase active site residues were analysed using Discovery Studio Visualizer. The interaction patterns for each compound are presented in Table 2, and the two-dimensional interaction diagrams are shown in Figures 3 to 9.
|
Compound |
Conventional Hydrogen Bond Interactions |
Carbon Hydrogen Bond / Other H-Bond Interactions |
Hydrophobic / Pi Interactions |
Other Important Interactions |
|
QD-1 (Mol 1) |
ARG B:54, GLU B:162, ARG B:53 |
GLY B:38 |
PRO B:50, ILE B:348 |
Halogen interaction with GLU B:43; unfavourable donor-donor interaction observed |
|
QD-2 (Mol 2) |
ASP B:305 |
THR B:230 |
PRO B:119, TRP B:103, LEU B:312 |
Pi-Pi T-shaped interaction with TRP B:103 |
|
QD-3 (Mol 3) |
ARG B:75 |
ARG A:75, GLY B:82 |
GLU A:79, GLU B:79 |
Pi-anion interactions with GLU A:79 and GLU B:79; unfavourable interaction with HOH A:550 |
|
QD-4 (Mol 4) |
ASN A:279, ARG A:98 |
GLY A:120, ASP A:122, GLN A:101 |
PRO A:124, VAL A:97 |
Halogen interaction with ASP A:94; unfavourable donor-donor interaction with TRP A:103 |
|
QD-5 (Mol 5) |
ASN A:279, ARG A:98 |
GLY A:120 |
PRO A:124, VAL A:97 |
Halogen interaction with ASP A:94; unfavourable interactions with HOH A:610 and TRP A:103 |
|
QD-6 (Mol 6) |
ARG B:75 |
ASN B:83 |
GLU A:79, GLU B:79 |
Pi-anion interactions with GLU A:79 and GLU B:79 |
|
Ciprofloxacin |
- |
Water-mediated interaction with HOH A:590 and HOH A:518 |
ILE A:36 |
Halogen interaction with TYR A:28; alkyl interaction with ALA A:32; unfavourable interactions with TYR A:31 and VAL A:35 |
Table 2. Amino Acid Interactions of Designed Quinolone Derivatives with DNA Gyrase (PDB: 3IFZ)
QD-1 formed three conventional hydrogen bonds with ARG B:54, GLU B:162, and ARG B:53, along with a carbon-hydrogen bond with GLY B:38. Hydrophobic contacts were observed with PRO B:50 and ILE B:348. A halogen interaction with GLU B:43 was also noted, though an unfavourable donor-donor interaction was observed in the binding pocket. QD-2 formed a conventional hydrogen bond with ASP B:305 and a carbon-hydrogen bond with THR B:230. Hydrophobic and pi interactions with PRO B:119, TRP B:103, and LEU B:312 were evident, along with a notable Pi-Pi T-shaped interaction with TRP B:103. QD-3 showed hydrogen bonding with ARG B:75 and ARG A:75, GLY B:82, with pi-anion interactions involving GLU A:79 and GLU B:79. An unfavourable contact with HOH A:550 was observed. QD-4 formed two conventional hydrogen bonds with ASN A:279 and ARG A:98, with additional carbon-hydrogen bonds involving GLY A:120, ASP A:122, and GLN A:101. Hydrophobic contacts with PRO A:124 and VAL A:97 were seen, and a halogen interaction with ASP A:94 was detected alongside an unfavourable contact with TRP A:103.
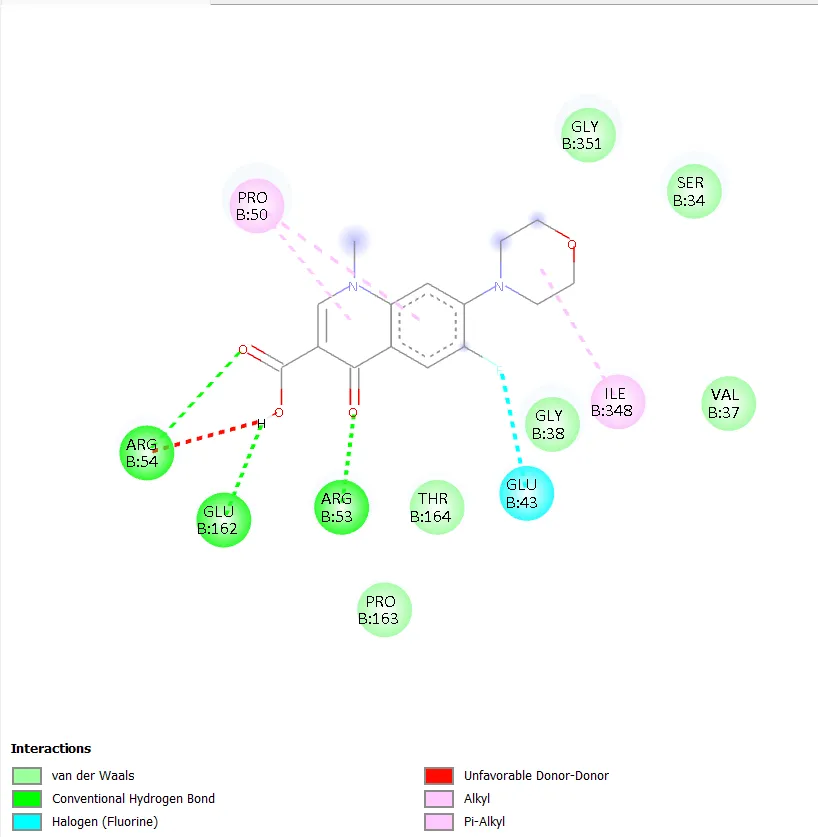
Figure 3. Two-dimensional protein-ligand interaction diagram of QD-1 with DNA gyrase.
|
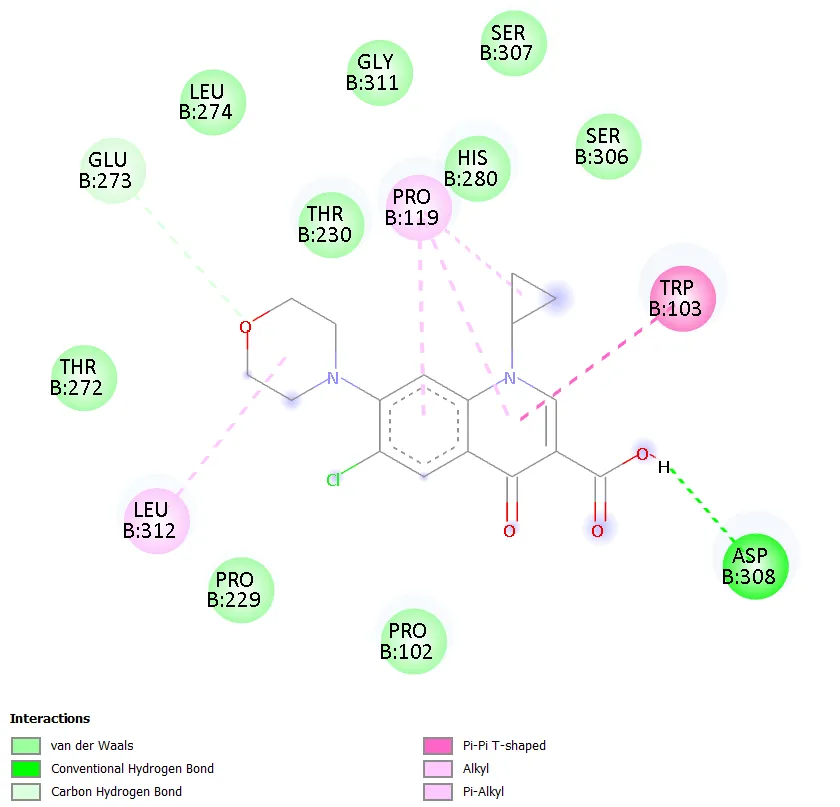
Figure 4. Two-dimensional protein-ligand interaction diagram of QD-2 with DNA gyrase. |
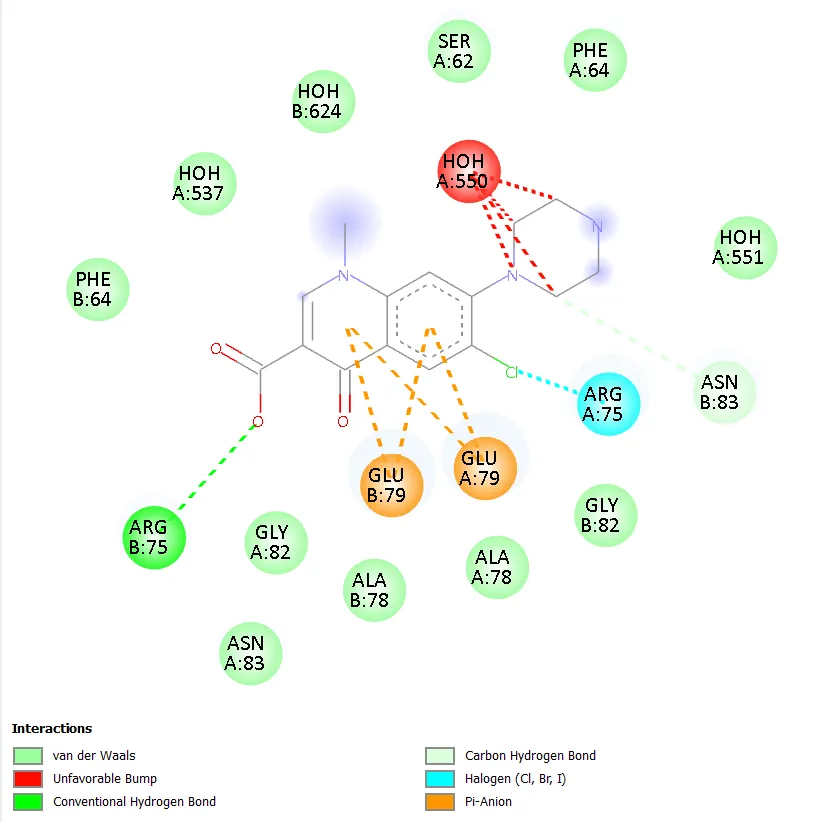
Figure 5. Two-dimensional protein-ligand interaction diagram of QD-3 with DNA gyrase. |
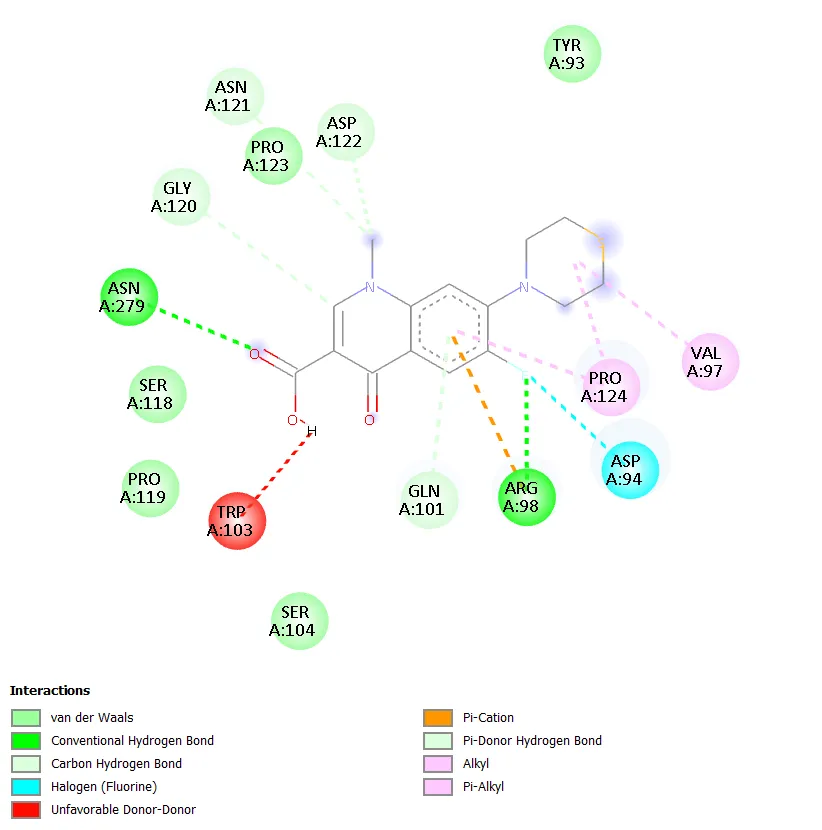
Figure 6. Two-dimensional protein-ligand interaction diagram of QD-4 with DNA gyrase. |
QD-5 formed two conventional hydrogen bonds with ASN A:279 and ARG A:98, mirroring the key binding interactions of QD-4. A carbon-hydrogen bond with GLY A:120 was also observed. Hydrophobic contacts with PRO A:124 and VAL A:97 were detected, along with a halogen interaction with ASP A:94. Unfavourable interactions with HOH A:610 and TRP A:103 were noted, though these did not prevent strong overall binding. The combination of multiple conventional hydrogen bonds, hydrophobic contacts, and a halogen interaction in QD-5 is consistent with its superior docking score of -7.4 kcal/mol. QD-6 showed hydrogen bonding with ARG B:75 and a carbon-hydrogen bond with ASN B:83, with pi-anion interactions involving GLU A:79 and GLU B:79, similar to QD-3. Ciprofloxacin, in contrast, did not show any clearly defined conventional hydrogen bonds but exhibited water-mediated interactions through HOH A:590 and HOH A:518, a hydrophobic contact with ILE A:36, a halogen interaction with TYR A:28, and an alkyl interaction with ALA A:32.
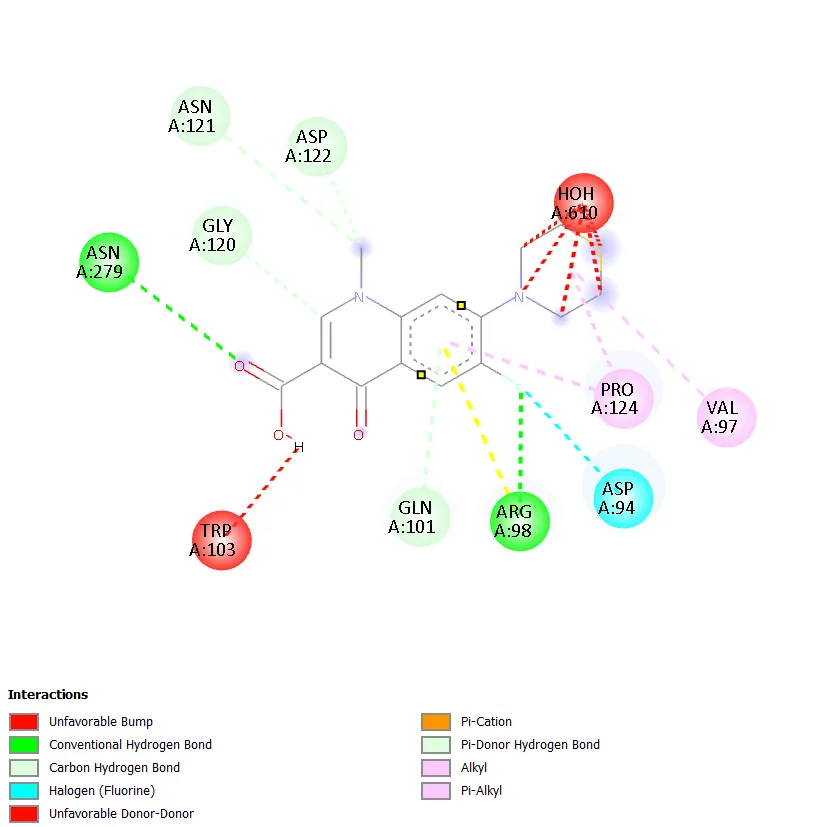
Figure 7. Two-dimensional protein-ligand interaction diagram of QD-5 with DNA gyrase. |
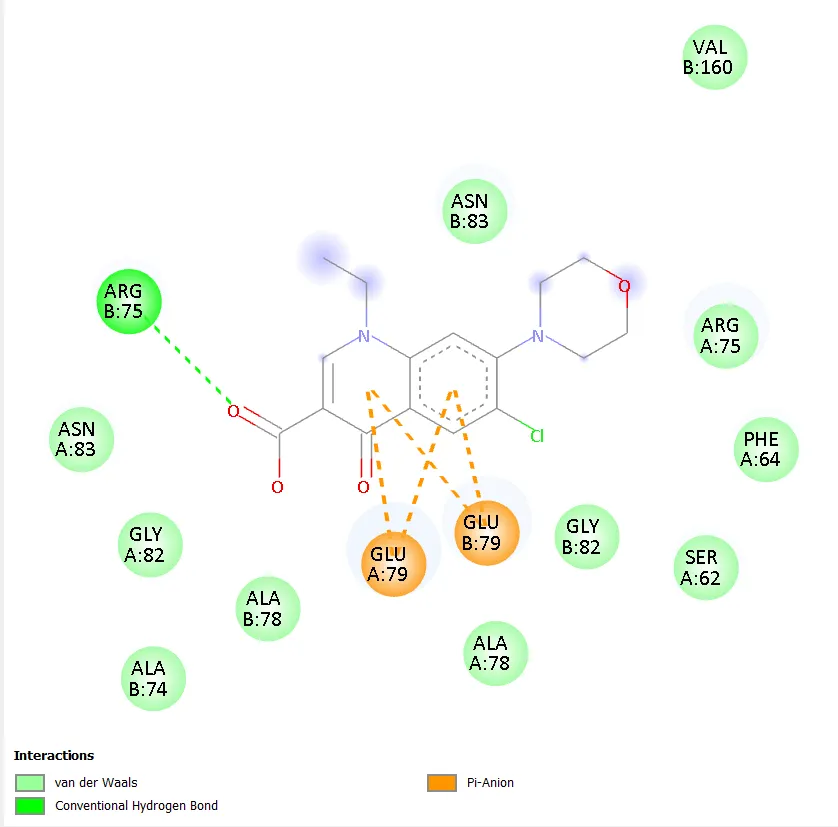
Figure 8. Two-dimensional protein-ligand interaction diagram of QD-6 with DNA gyrase. |
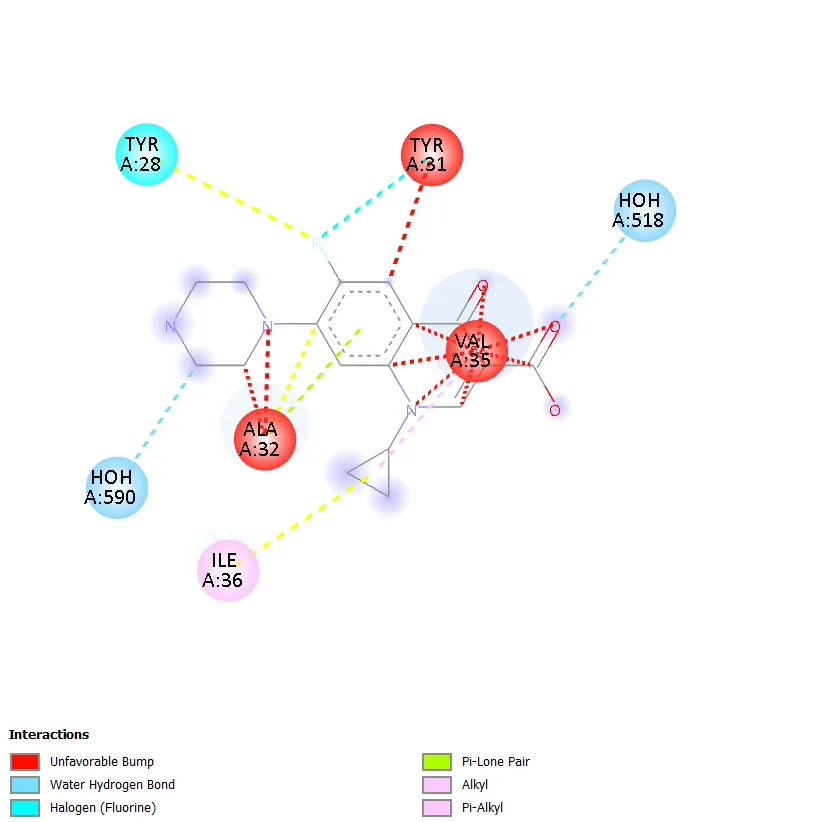
Figure 9. Two-dimensional protein-ligand interaction diagram of Ciprofloxacin with DNA gyrase. |
|
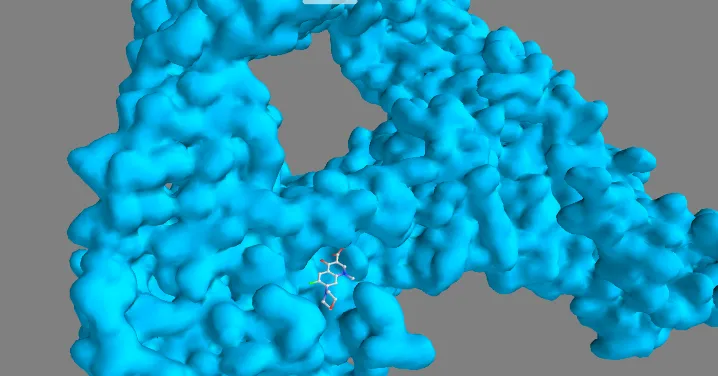
Figure 10. Three-dimensional docking binding pose of QD-5 within the active site of DNA gyrase.

Figure 11. Three-dimensional docking binding pose of Ciprofloxacin within the active site of DNA gyrase.
4.4 ADMET and Drug-Likeness Properties
The pharmacokinetic and drug-likeness properties of all designed compounds were evaluated using SwissADME and are summarised in Table 3.
|
Compound |
TPSA (Ų) |
HBA |
HBD |
RB |
LogP |
BBB |
Lipinski |
Bioavailability Score |
|
QD-1 |
74.57 |
5 |
2 |
2 |
0.53 |
No |
0 violation |
0.55 |
|
QD-2 |
71.77 |
5 |
1 |
3 |
1.78 |
Yes |
0 violation |
0.85 |
|
QD-3 |
65.78 |
5 |
1 |
3 |
1.36 |
Yes |
0 violation |
0.55 |
|
QD-4 |
88.56 |
6 |
2 |
3 |
1.34 |
No |
0 violation |
0.55 |
|
QD-5 |
94.80 |
5 |
3 |
3 |
0.96 |
No |
0 violation |
0.55 |
|
QD-6 |
133.66 |
6 |
1 |
4 |
1.66 |
No |
0 violation |
0.56 |
|
Ciprofloxacin |
74.57* |
6 |
2 |
1 |
0.28* |
No |
0 violation |
0.55* |
Table 3: ADME and Drug-Likeness Properties of Designed Quinolone Derivatives
Abbreviations: TPSA = Topological Polar Surface Area; HBA = Hydrogen Bond Acceptors; HBD = Hydrogen Bond Donors; RB = Rotatable Bonds; BBB = Blood-Brain Barrier; *Values for ciprofloxacin are from standard reference data.
All six quinolone derivatives had molecular weights below 500 g/mol, satisfying the first criterion of Lipinski's Rule of Five [10]. The topological polar surface area (TPSA) values ranged from 65.78 A2 (QD-3) to 133.66 A2 (QD-6), with values below 140 A2 being generally associated with adequate oral absorption. All compounds demonstrated high gastrointestinal (GI) absorption, which is a critical indicator of oral bioavailability. QD-2 and QD-3 showed blood-brain barrier (BBB) penetration, while the remaining compounds did not cross the BBB, which may be pharmacologically advantageous for antibacterial agents where central nervous system (CNS) penetration is not required. All compounds showed zero violations of Lipinski's Rule of Five, indicating that they possess acceptable drug-likeness characteristics. QD-2 had the highest bioavailability score (0.85), while most other compounds scored 0.55 to 0.56. The consensus Log P values of all compounds were within the acceptable range (below 5), indicating suitable lipophilicity for oral drug candidates. QD-5 had a consensus Log P of 0.96 and a TPSA of 94.80 A2, both within the acceptable range, further supporting its candidacy as a lead compound.
4.5 ProTox-II Toxicity Prediction
The predicted toxicity profiles of the designed compounds, including acute oral LD50 values, GHS toxicity classes, and organ-specific toxicity endpoints, are presented in Table 4. All compounds were classified into GHS toxicity class IV (LD50 between 300 and 2000 mg/kg) or class V (LD50 between 2000 and 5000 mg/kg). QD-1 and QD-2 had predicted LD50 values of 3000 mg/kg (class V), indicating relatively low acute oral toxicity. QD-3 and QD-5 had LD50 values of 2000 mg/kg (class IV), while QD-4 and QD-6 showed 2500 mg/kg (class IV and V respectively). Ciprofloxacin itself predicted an LD50 of 2000 mg/kg and was placed in toxicity class IV, providing a useful reference point.
In terms of organ-specific toxicity, none of the designed compounds were predicted to show hepatotoxicity (liver toxicity) or cardiotoxicity, which is an important safety criterion. All compounds were predicted to be active for neurotoxicity, nephrotoxicity, and respiratory toxicity, though these predictions are computational estimations that require experimental confirmation. Carcinogenicity, immunotoxicity, and cytotoxicity were all predicted as inactive for all six compounds. Mutagenicity was predicted as active for QD-3, QD-4, and QD-5, which also matched the mutagenicity prediction for ciprofloxacin itself. The overall toxicity profiles of the designed compounds were broadly comparable to that of the reference drug, suggesting an acceptable in silico safety profile.
|
Compound |
LD50 (mg/kg) |
Toxicity Class |
Neurotoxicity |
Nephrotoxicity |
Respiratory Toxicity |
Mutagenicity |
|
QD-1 |
3000 |
V |
Active |
Active |
Active |
Inactive |
|
QD-2 |
3000 |
V |
Active |
Active |
Active |
Inactive |
|
QD-3 |
2000 |
IV |
Active |
Active |
Active |
Active |
|
QD-4 |
2500 |
IV |
Active |
Active |
Active |
Active |
|
QD-5 |
2000 |
IV |
Active |
Active |
Active |
Active |
|
QD-6 |
2500 |
V |
Active |
Active |
Active |
Inactive |
|
*Ciprofloxacin |
2000 |
IV |
Active |
Active |
Active |
Active |
Abbreviations: LD50 = Lethal dose 50 (mg/kg); Tox Class = GHS Toxicity Class (IV = harmful if swallowed, V = may be harmful if swallowed). Active/Inactive represent predicted endpoint activity.
5. DISCUSSION
The present study describes an in-silico approach to evaluating six designed quinolone derivatives for their potential as antibacterial agents targeting DNA gyrase of M. tuberculosis. The molecular docking, ADMET profiling, and toxicity prediction results collectively provide a useful basis for identifying promising lead compounds from within this designed series.
Binding energy, expressed as the docking score in kcal/mol, is a direct indicator of the predicted binding affinity between a ligand and its target protein. A more negative docking score corresponds to a more stable and energetically favourable ligand-protein complex, and thus a higher predicted affinity. Among the six designed compounds, QD-5 produced the best docking score of -7.4 kcal/mol, which surpassed that of the reference drug ciprofloxacin (-7.3 kcal/mol). QD-2 matched ciprofloxacin at -7.3 kcal/mol, while QD-4 (-7.2 kcal/mol) and QD-1 (-7.1 kcal/mol) also showed competitive binding affinities. QD-6 (-6.8 kcal/mol) and QD-3 (-6.7 kcal/mol) showed lower affinities. The fact that QD-5 surpassed ciprofloxacin in predicted binding affinity is encouraging and suggests that the structural features of QD-5 are well suited for interaction with the DNA gyrase active site.
QD-5 carries a cyclopropyl group at N-1, a methoxy substituent at C-6, and a piperazine ring at C-7. The cyclopropyl group at N-1 is a structural feature also present in ciprofloxacin and is known to enhance antibacterial activity by improving binding interactions within the gyrase active site [2,3]. The methoxy group at C-6 is a polar substituent that can participate in hydrogen bonding or electrostatic interactions with active site residues. The piperazine ring at C-7 provides a nitrogen atom capable of ionic and hydrogen bonding interactions, which are known to be important for quinolone pharmacological activity [4]. This combination of substituents appears to have produced a compound with an optimal fit within the active site of DNA gyrase. Interaction analysis showed that QD-5 formed two conventional hydrogen bonds with ASN A:279 and ARG A:98, hydrophobic contacts with PRO A:124 and VAL A:97, and a halogen interaction with ASP A:94. These multiple points of contact collectively stabilise the ligand within the binding pocket and explain the favourable docking score.
The importance of hydrogen bonding in ligand-receptor interaction cannot be overstated. Hydrogen bonds provide directionality and specificity to ligand binding, contributing significantly to binding affinity and selectivity [5]. QD-1 formed the largest number of conventional hydrogen bonds in this series (three bonds with ARG B:54, GLU B:162, and ARG B:53), which likely accounts for its reasonably good docking score of -7.1 kcal/mol. QD-4 shared the same key binding residues (ASN A:279 and ARG A:98) as QD-5 but with additional carbon-hydrogen bonds and a slightly lower docking score, indicating that the specific nature of the substituents also modulates the quality of the binding interactions. QD-2 demonstrated a unique Pi-Pi T-shaped interaction with TRP B:103, which is a type of aromatic ring stacking interaction that can provide significant stabilisation, consistent with its equal docking score to ciprofloxacin. Hydrophobic interactions involving residues such as PRO, ILE, LEU, and VAL contribute to stabilising the ligand within non-polar regions of the binding pocket through van der Waals forces. These interactions, while individually weaker than hydrogen bonds, cumulatively contribute to binding affinity [5,6].
The ADMET analysis confirms that all six compounds are likely to be orally bioavailable and are predicted to be well absorbed through the gastrointestinal tract. All compounds satisfied Lipinski's Rule of Five, which sets threshold values of MW below 500, Log P below 5, HBD below 5, and HBA below 10 for oral drug candidates [10]. QD-5, with a MW of 363.80, Log P of 0.96, TPSA of 94.80 A2, and high GI absorption, shows favourable pharmacokinetics. The lack of BBB penetration in QD-5 is consistent with its intended use as a peripherally acting antibacterial agent and is not considered a liability in this context. The relatively low consensus Log P value of QD-5 (0.96) compared to ciprofloxacin (0.28) suggests slightly higher lipophilicity, which may contribute to better tissue penetration and intracellular activity against M. tuberculosis, a pathogen that primarily resides within host macrophages [1].
The ProTox-II toxicity predictions indicate that QD-5 belongs to GHS toxicity class IV with a predicted LD50 of 2000 mg/kg, which is comparable to ciprofloxacin (also class IV, LD50 2000 mg/kg). The predicted absence of hepatotoxicity and cardiotoxicity in QD-5 is particularly noteworthy, as these are among the most serious adverse effects that have led to the withdrawal or dose restriction of several antibacterial drugs in clinical practice. The predicted mutagenicity of QD-5 is a flag that warrants experimental investigation; however, it is worth noting that ciprofloxacin itself carries the same mutagenicity prediction in this model, which should be interpreted with caution given that ciprofloxacin is widely used clinically and its mutagenic risk in clinical settings is considered low at therapeutic doses. These computational predictions are preliminary and require confirmation through experimental assays such as the Ames test and cytotoxicity assays in cell lines.
The present study has certain limitations that are inherent to the in-silico methodology. Molecular docking scores predict binding affinity but do not account for protein flexibility, solvent effects, entropy contributions to binding, or the complex biological environment within living cells. ADMET predictions are based on computational models trained on known drug datasets and may not perfectly capture the properties of structurally novel compounds. Similarly, ProTox-II predictions are based on structural similarity to known toxic compounds and carry inherent uncertainty for novel scaffolds. Despite these limitations, computational approaches represent a valuable first step in drug discovery, providing rational guidance for the selection of candidates to advance to experimental validation. The findings of this study justify the synthesis of QD-5 and related compounds followed by in vitro antibacterial activity testing, cytotoxicity assays, and, if promising results are obtained, in vivo pharmacological studies.
6. FUTURE SCOPE
The present in silico findings provide preliminary evidence supporting the antibacterial potential of the designed quinolone derivatives, particularly QD-5. Future research should focus on the chemical synthesis of QD-5 and the other promising derivatives (QD-1 and QD-2) followed by their in vitro antibacterial activity evaluation against M. tuberculosis strains, including drug-susceptible and drug-resistant strains, using minimum inhibitory concentration (MIC) assays. In vitro cytotoxicity studies in human cell lines should be performed to assess safety at the cellular level. Molecular dynamics simulation studies can further evaluate the stability of the QD-5 and DNA gyrase complex over time and provide a deeper understanding of binding dynamics. Optimisation of the QD-5 scaffold through further structural modifications, followed by structure-activity relationship (SAR) analysis, may lead to compounds with even greater potency and improved selectivity. In vivo pharmacological and pharmacokinetic studies in animal models will ultimately be required to evaluate efficacy, safety, and drug metabolism before clinical translation can be considered.
CONCLUSION
This study successfully designed and computationally evaluated six novel quinolone derivatives as potential antibacterial agents against DNA gyrase of Mycobacterium tuberculosis using molecular docking, ADMET prediction, and toxicity profiling. Among the six designed compounds, QD-5 (1-cyclopropyl-6-methoxy-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid) demonstrated the highest binding affinity against DNA gyrase with a docking score of -7.4 kcal/mol, which was better than the reference drug ciprofloxacin (-7.3 kcal/mol). Interaction analysis revealed that QD-5 engages key active site residues including ASN A:279 and ARG A:98 through conventional hydrogen bonds, along with hydrophobic contacts and a halogen interaction, collectively stabilising the ligand within the binding pocket. All designed compounds satisfied Lipinski's Rule of Five and showed high gastrointestinal absorption, confirming their drug-likeness. ProTox-II predictions indicated acceptable acute oral toxicity profiles, with no predicted hepatotoxicity or cardiotoxicity for any compound. Based on the combined results of molecular docking, ADMET analysis, and toxicity prediction, QD-5 emerges as the most promising lead compound in this series for potential development as an antibacterial agent against M. tuberculosis. These computational findings provide a rational foundation for future experimental validation of these compounds.
REFERENCES




Manoj Gangadhar Shinde, Akshat Jitendra Bhamare*, Janhavi Harish Bhalerao, Tejal Hari Bare, Rutuja Purushottam Bhojane, In Silico Computational Drug Design On Quinolone Derivatives As Antibacterial Agents, Int. J. Sci. R. Tech., 2026, 3 (6), 259-272. https://doi.org/10.5281/zenodo.20538244
 10.5281/zenodo.20538244
10.5281/zenodo.20538244