
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
R.G Sapkal College of Pharmacy, Sapkal Knowledge Hub, Kalyani Hills, Anjaneri, Trimbakeshwar Rd, Nashik, 422213, Maharashtra, India.
Prucalopride succinate is a first-in-class, highly selective serotonin 5-HT4 receptor agonist used for the symptomatic treatment of chronic idiopathic constipation in adults who have not achieved adequate relief with laxatives. As with any active pharmaceutical ingredient, reliable quantification of prucalopride in bulk drug and in finished dosage forms, together with a thorough understanding of its degradation behaviour, is essential for ensuring product quality, safety and regulatory compliance throughout the product life cycle. This review critically examines the application of the Quality by Design (QbD) philosophy to the development of reverse-phase high-performance liquid chromatography (RP-HPLC) methods for prucalopride, and consolidates the available literature on stability-indicating, forced degradation-based assay methods for the drug. The QbD framework, anchored in the Analytical Target Profile, Critical Analytical Attributes, risk assessment tools and Design of Experiments (DoE) strategies such as Plackett-Burman screening and Box-Behnken/Central Composite optimization designs, is discussed in the context of building method robustness directly into the chromatographic conditions rather than discovering it after the fact. The review further compiles the reported stress-degradation behaviour of prucalopride under acidic, alkaline, oxidative, thermal, photolytic and neutral hydrolytic conditions, and evaluates how peak purity and mass-balance data have been used to establish specificity of the reported assays. Validation parameters specificity, linearity, accuracy, precision, LOD/LOQ, robustness and system suitability reported across the available literature are summarised and benchmarked against ICH Q2(R1)/Q2(R2) and Q1A(R2) expectations. A comparative table of UV-spectrophotometric, RP-HPLC, LC-MS/MS and other reported methods for prucalopride is provided, along with a discussion of the practical advantages that a QbD-driven, stability-indicating RP-HPLC method offers over conventional one-factor-at-a-time (OFAT) approaches. The review concludes that combining analytical QbD principles with a systematic forced degradation study offers a scientifically sound, regulatorily robust, and economically efficient pathway for developing a validated stability-indicating method for prucalopride suitable for routine quality control, stability studies and regulatory submission.
1.1 Prucalopride: Pharmacological Profile and Receptor Selectivity
Prucalopride, administered clinically as its succinate salt, is a dihydro-benzofuran-carboxamide derivative and represents a first-in-class, highly selective, high-affinity agonist of the serotonin type-4 (5-HT4) receptor1,2,3. Unlike earlier generation enterokinetic agents such as cisapride and tegaserod, which showed clinically significant off-target activity at cardiac hERG channels and at 5-HT1B/1D receptors respectively, prucalopride demonstrates more than 150-fold selectivity for the 5-HT4 receptor over other receptor subtypes, a property that was deliberately engineered during its rational drug-design process to avoid the cardiovascular liabilities that led to withdrawal of its predecessors4,5,6. Activation of 5-HT4 receptors located on enteric neurons of the gastrointestinal tract enhances the release of acetylcholine, thereby stimulating smooth-muscle peristalsis, improving coordination between the circular and longitudinal muscle layers of the colon, and accelerating colonic transit7. Multiple randomized, placebo-controlled trials have confirmed the efficacy of prucalopride in patients with chronic idiopathic constipation who have failed conventional laxative therapy, with benefits sustained over long-term, open-label follow-up 1,2. Regulatory approval has since been granted in numerous jurisdictions, with the drug marketed globally under brand names including Motegrity, Resolor and Resotran 8.
1.2 Physicochemical Properties and pH-Dependent Behaviour
Prucalopride succinate is chemically described as 4-amino-5-chloro-2,3-dihydro-N-[1-(3-methoxypropyl)-4-piperidinyl]-7-benzofurancarboxamide succinate, with a molecular weight of 485.96 g/mol for the succinate salt 6. The drug substance is a white to off-white crystalline powder with a melting point of approximately 198 °C. It is freely soluble in acidic aqueous media, sparingly soluble in methanol, and readily soluble in polar aprotic solvents such as N,N-dimethylformamide, dimethylsulfoxide and N,N-dimethylacetamide, but its aqueous solubility decreases markedly with increasing pH 6. The molecule possesses two ionisable centres: the piperidine nitrogen, with a reported pKa of approximately 8.5, and the aromatic primary amino group, with a pKa of less than 3, both determined at 20 °C 6. This pH-dependent ionisation behaviour has direct consequences for RP-HPLC method development, since retention, peak shape and resolution of prucalopride are all sensitive to the pH of the aqueous mobile-phase component, making buffer pH one of the most critical method parameters to control and optimize.
Figure 1: Chemical Structure and Pharmacological Mechanism of Prucalopride
1.3 Pharmacokinetic Profile
Following oral administration, prucalopride is rapidly absorbed, is minimally metabolised, and is predominantly excreted unchanged in the urine, giving it a relatively simple and predictable pharmacokinetic profile that supports once-daily dosing 4,8. Its pharmacodynamic action is restricted essentially to enhancement of colonic motility, with a well-characterised, generally mild and self-limiting side-effect profile most notable at treatment initiation 8. This pharmacological simplicity contrasts with the analytical complexity introduced by the drug's dual ionisable centres and its susceptibility to degradation, which together necessitate a carefully optimized and rigorously validated chromatographic method.
1.4 Marketed Formulation and Analytical Requirements
Prucalopride succinate is marketed as immediate-release film-coated tablets, typically in 1 mg and 2 mg strengths, intended for once-daily oral administration 6. As with all solid oral dosage forms, the marketed product must be supported by validated analytical methods capable of confirming identity, assay/potency, content uniformity, dissolution performance and stability throughout shelf life, in the presence of tablet excipients and any degradation products that may arise on storage.
1.5 Importance of Systematic Analytical Method Development
Analytical method development is not a peripheral activity in pharmaceutical quality assurance; it is the backbone upon which every downstream quality decision batch release, stability commitment, and regulatory filing ultimately rests. A method that has not been systematically developed and thoroughly validated risks reporting inaccurate assay values, missing genuine degradation products, or giving false confidence in a formulation's stability. For a molecule such as prucalopride, whose retention behaviour is pH-dependent and for which limited compendial guidance exists (prucalopride does not yet carry a monograph in the major pharmacopoeias), method developers must build a chromatographic method essentially from first principles. This makes prucalopride a particularly instructive case study for demonstrating how a systematic, risk-based QbD approach can be applied in preference to traditional trial-and-error (OFAT) method development.
1.6 Regulatory Requirement for Stability-Indicating Methods
Regulatory agencies including the US FDA, EMA and ICH member authorities require that assay and related-substance methods used for drug substance and drug product release and stability testing be demonstrably "stability-indicating" that is, capable of accurately quantifying the intact drug in the presence of process-related impurities and degradation products without positive or negative interference 33,35. ICH Q1A(R2) mandates that stress (forced degradation) testing be performed on new drug substances to establish degradation pathways and to confirm that the analytical procedure used for stability studies is suitably specific 33. A method that has not been challenged against acid, base, oxidative, thermal, photolytic and humidity stress cannot be assumed to be free from co-elution artefacts, and therefore cannot be relied upon for shelf-life determination or out-of-specification investigations.
1.7 Objectives of this Review
The objective of this review is threefold: first, to consolidate the principles of analytical Quality by Design and to describe systematically how these principles have been, and can be, applied to the development of an RP-HPLC method for prucalopride; second, to compile and critically evaluate the forced degradation and stability-indicating method literature reported for prucalopride, including degradation pathways, stress conditions and mass-balance considerations; and third, to benchmark the QbD-optimized stability-indicating RP-HPLC approach against other reported analytical techniques (UV-spectrophotometric, fluorimetric, electrochemical and LC-MS/MS) for prucalopride, so as to provide method developers, quality control chemists and regulatory reviewers with a single, structured reference document on the subject.
2. QUALITY BY DESIGN (QbD) IN ANALYTICAL METHOD DEVELOPMENT:
2.1 Principles of Analytical Quality by Design
Quality by Design (QbD) is a systematic, science- and risk-based approach to product and process development that begins with predefined objectives and emphasises understanding of the relationship between input variables and output quality 37,38. Although QbD was originally conceived by ICH Q8(R2) for pharmaceutical product and process development, the same underlying philosophy proactively building quality into a system rather than testing for it afterward has since been extended to analytical procedures, giving rise to the discipline now widely termed Analytical Quality by Design (AQbD). ICH Q14, issued alongside the revised Q2(R2) guideline, formally codifies analytical procedure development within this risk-based framework, providing regulatory recognition for AQbD-derived methods and for the concept of a defined Method Operable Design Region 36.
2.2 Analytical Target Profile and Critical Analytical Attributes
The starting point of any AQbD exercise is the definition of an Analytical Target Profile (ATP), a prospective summary of the performance characteristics that the finished analytical method must deliver for example, the required specificity toward degradation products, the linearity range needed to cover 50-150% of label claim, and the precision and accuracy limits acceptable for batch release. From the ATP, Critical Analytical Attributes (CAAs) are identified: measurable method outputs such as resolution between the prucalopride peak and its nearest degradation product, tailing factor, theoretical plate count and retention time, whose values directly determine whether the ATP is met. Critical Method Parameters (CMPs) typically mobile-phase pH, organic modifier ratio, buffer concentration, flow rate, column temperature and detection wavelength are the input variables that the analyst can control and that are known or suspected to influence the CAAs18.
2.3 Risk Assessment
Risk assessment is used to narrow the universe of potential method parameters down to those genuinely likely to affect method performance, before committing to a full experimental optimization study. An Ishikawa (fishbone) diagram is commonly constructed with the CAA (for example, "resolution and peak symmetry of prucalopride") as the effect, and instrument-, column-, mobile-phase-, and procedure-related factors as the contributing causes. A risk-ranking and filtering exercise, or a simple risk matrix scoring severity and likelihood, is then used to shortlist the parameters carried forward into formal screening. For prucalopride, mobile-phase pH is consistently ranked as a high-risk parameter because of the drug's two ionisable functional groups, together with buffer concentration and the ratio of organic modifier, while parameters such as injection volume or needle wash solvent are typically ranked low risk and are fixed rather than formally optimized19.
2.4 Design of Experiments (DoE)
Design of Experiments (DoE) provides the statistical backbone of AQbD-based method development, replacing the traditional one-factor-at-a-time (OFAT) approach in which each parameter is varied independently while all others are held constant. OFAT is inefficient, requires a disproportionately large number of experimental runs, and critically cannot detect interaction effects between parameters, which are frequently the dominant source of chromatographic variability20. DoE strategies are typically applied in two stages. Screening designs, such as the two-level Plackett-Burman or fractional factorial design, are used first to evaluate a relatively large number of candidate factors (mobile-phase pH, buffer strength, flow rate, column temperature, wavelength, injection volume) in a small number of experimental runs, in order to statistically identify which factors have a significant effect on the CAAs and which can be fixed at a convenient level. Optimization designs are then applied to the significant factors identified at the screening stage. The Box-Behnken design (BBD) and Central Composite Design (CCD) are the most widely reported response-surface methodologies in the pharmaceutical RP-HPLC literature, each evaluating three factors at three levels in a limited number of runs while allowing quadratic and two-factor interaction terms to be modelled21.
2.5 Response Surface Methodology and Design Space
Once response-surface models have been generated for each CAA (retention time, resolution, tailing factor, theoretical plates, peak area, run time) as a function of the CMPs, contour plots and three-dimensional response-surface plots are used to visualise the design space graphically. Statistical software such as Design-Expert or Minitab is typically used to fit polynomial models, assess model significance by ANOVA, and generate overlay plots that superimpose the acceptable ranges for each CAA, allowing identification of a combined region the Design Space, or in analytical terms the Method Operable Design Region (MODR) within which all CAAs simultaneously meet their target criteria. A set of chromatographic conditions is then selected from within this region, typically at or near a point of maximum desirability, and this "optimized" condition becomes the proposed method, which is subsequently subjected to full ICH Q2(R1)/Q2(R2) validation22.
2.6 Method Operable Design Region (MODR)
A defining feature of the MODR concept is that, provided the method is operated with any combination of parameters lying within the design space, method performance is assured without the need for re-validation a concept directly analogous to the "design space" concept in product and process QbD under ICH Q8(R2). This has practical regulatory value: a laboratory can make small, justified adjustments to a validated CMP (for example, a small pH or flow-rate adjustment during routine analysis to compensate for column-to-column variability) without triggering a full re-validation, provided the adjustment remains within the established MODR23.
2.7 Control Strategy and Lifecycle Management
Control strategy is the final pillar of AQbD, encompassing system suitability criteria, specified control limits on CMPs, and a documented plan for how the method will be monitored and, where needed, improved over its life cycle. This is philosophically distinct from the traditional validate-once approach; QbD treats method validation as the beginning of a continuous verification process rather than a one-time gate24.
2.8 Advantages of AQbD over OFAT Approaches
Compared with traditional OFAT method development, the AQbD approach offers several well-documented advantages: a smaller total number of experimental runs for a comparable or greater amount of information gained; explicit quantification of interaction effects between mobile-phase pH, organic ratio and flow rate, which are frequently significant for basic, ionisable analytes such as prucalopride; a statistically justified design space that supports post-approval flexibility; and, ultimately, a more robust method less prone to failure during transfer between laboratories, instruments or analysts 17,18. These advantages have been repeatedly demonstrated for other small-molecule APIs including evogliptin tartrate, prasugrel hydrochloride, palovarotene, imeglimin hydrochloride, tadalafil, and the fixed-dose combination of rosuvastatin and clopidogrel, in each case using Box-Behnken designs to optimize mobile-phase pH, organic modifier ratio and flow rate as the critical method parameters25.
2.9 AQbD Workflow Applied to Prucalopride RP-HPLC
Applied specifically to RP-HPLC method development, the AQbD workflow for a molecule such as prucalopride proceeds through definition of the ATP (assay and stability-indicating capability across 50-150% of label claim, resolution ≥ 2.0 from all known and unknown degradation products), risk assessment to shortlist mobile-phase pH, buffer concentration, acetonitrile/methanol ratio and flow rate as high-risk CMPs, Plackett-Burman screening to confirm significance, Box-Behnken or central composite optimization of the shortlisted factors against resolution, tailing factor, theoretical plates and retention time as CAAs, definition of the MODR, and selection of a robust operating point, which is then locked in as the proposed method and taken forward to formal validation and forced degradation challenge.
3. RP-HPLC METHOD DEVELOPMENT FOR PRUCALOPRIDE:
3.1 Instrument and Detector Selection
Selection of the analytical platform for prucalopride begins with the instrument and detector. Because prucalopride possesses a chromophore associated with its benzofuran-carboxamide nucleus, it exhibits useful UV absorbance, with reported absorption maxima in the region of 226 nm in acidic media and detection wavelengths of 276-277 nm commonly employed for HPLC quantification once the analyte is separated from matrix and mobile-phase background absorbance 14,31. A conventional HPLC system fitted with a UV or, preferably, a photodiode-array (PDA) detector is therefore adequate and is the platform most consistently reported in the literature; PDA detection carries the additional advantage of enabling peak-purity assessment, which is essential for establishing specificity during forced degradation studies.
3.2 Stationary Phase Selection
Column (stationary phase) selection is one of the earliest and most consequential decisions in method development. Reported RP-HPLC methods for prucalopride have employed octadecylsilane (C18) and octylsilane (C8) bonded-phase columns of conventional dimensions (150-250 mm length, 4.6 mm internal diameter, 5 μm particle size), including Grace C18, Symmetry C8 and Hypersil-type columns, as well as sub-2-micron columns for UPLC method transfer 9,10. A C18 phase is generally the default starting point for a moderately polar, ionisable basic molecule such as prucalopride because it offers a good balance of retention, selectivity and peak shape for basic analytes when combined with an appropriately buffered, acidic mobile phase; C8 phases have also been reported to provide adequate retention with somewhat reduced analysis time.
3.3 Mobile Phase Optimization
Mobile-phase optimization is the single most influential step in developing a method for prucalopride, given the compound's two ionisable centres (piperidine pKa ≈ 8.5; aromatic amine pKa < 3). Reported mobile phases are predominantly composed of a buffered aqueous component potassium dihydrogen phosphate, ammonium formate, or triethylamine-modified water adjusted to an acidic pH (pH 2.8-3.0) combined with acetonitrile as the organic modifier, typically in ratios ranging from approximately 80:20 to 82:18 (aqueous:organic, v/v) under isocratic conditions 9,10. Operating at low pH ensures that the basic piperidine nitrogen is fully protonated, giving reproducible, sharp, symmetrical peaks and minimising peak tailing that would otherwise arise from secondary interactions between the ionised amine and residual silanol groups on the stationary phase. Within a QbD framework, mobile-phase pH, buffer concentration/strength, and the ratio of organic modifier to aqueous buffer are treated as the principal Critical Method Parameters requiring formal DoE-based optimization, because small changes in any of these three variables produce disproportionately large shifts in retention time, resolution and peak shape for an ionisable analyte such as prucalopride.
3.4 Flow Rate, Detection Wavelength and Injection Parameters
Flow rate is typically optimized in the range of 0.8-1.0 mL/min for conventional 4.6 mm i.d. columns, balancing analysis time against backpressure and resolution; detection wavelength is set in the region of 225-277 nm depending on the mobile-phase composition and interfering matrix background, with 276-277 nm frequently preferred for tablet-formulation assays because it offers better selectivity from common excipient absorbance while retaining adequate analyte sensitivity5,9. Column temperature is commonly controlled at 25-30 °C to improve retention-time reproducibility, and injection volumes of 10-20 μL are typical for conventional HPLC-UV/PDA systems.
3.5 Box-Behnken Design for Method Optimization
Within the AQbD workflow, once mobile-phase pH, organic ratio and flow rate have been shortlisted through risk assessment and confirmed as significant by a screening design, a three-factor, three-level Box-Behnken design is the most frequently reported optimization tool for RP-HPLC methods of this type 18,19,20,21,22,23,24,25. A typical design matrix comprises 15-17 experimental runs, including replicated centre points, in which mobile-phase pH (for example, 2.5, 3.0, 3.5), acetonitrile content (for example, 15%, 18%, 21% v/v) and flow rate (for example, 0.8, 1.0, 1.2 mL/min) are varied systematically according to the BBD matrix, while retention time, resolution, tailing factor and theoretical plate count are recorded as responses for each run.
3.6 Response Surface Modelling
The response data generated from the design matrix are fitted to a quadratic polynomial model of the general form Y = β0 + β1X1 + β2X2 + β3X3 + β12X1X2 + β13X1X3 + β23X2X3 + β11X1² + β22X2² + β33X3², where Y represents each critical analytical attribute and X1, X2, X3 represent the coded values of pH, organic ratio and flow rate respectively. Analysis of variance (ANOVA) is used to test model significance (p < 0.05) and lack-of-fit, and the adequacy of the fitted model is further assessed using the coefficient of determination (R²) and predicted-versus-actual residual plots. Three-dimensional response-surface plots and two-dimensional contour plots are then generated for each CAA, allowing the analyst to visualise how resolution, tailing factor and retention time change jointly as functions of pH and organic ratio (or any other factor pair), at a fixed level of the third factor.
3.7 Definition of the Method Operable Design Region
Overlaying the individual contour plots for all CAAs of interest (for example, resolution ≥ 2.0, tailing factor ≤ 1.5, theoretical plates ≥ 2000, run time ≤ 10 minutes) defines the Method Operable Design Region the combined zone of the design space within which every critical attribute simultaneously satisfies its target criterion. A "sweet-spot" or point of maximum desirability is then selected from within this region, typically at or close to the centre of the MODR to maximise robustness margin against small, unavoidable day-to-day variation in mobile-phase preparation or instrument performance, and this combination of conditions is fixed as the final, optimized chromatographic method.
3.8 Optimized Chromatographic Conditions
Based on the pooled literature for prucalopride RP-HPLC methods, a representative optimized chromatographic condition consists of: C18 (or C8) column, 150-250 mm × 4.6 mm, 5 μm particle size; mobile phase of acetonitrile and phosphate or formate buffer (pH ~ 3.0) in the ratio of approximately 20:80 v/v; isocratic elution at a flow rate of 1.0 mL/min; column temperature of 30 °C; UV/PDA detection at 276-277 nm; and an injection volume of 10-20 μL, giving a retention time for prucalopride in the region of 5-6 minutes and a total run time of approximately 10 minutes 9. Under such conditions, system suitability parameters are consistently reported to meet compendial expectations: theoretical plate counts in excess of 2000, tailing factors below 1.5, and percentage RSD for replicate retention time and peak area determinations below 2%, confirming the suitability of the system for its intended quantitative purpose.
|
Parameter |
Reported Range / Typical Value |
|
Column |
C18 or C8, 150–250 mm × 4.6 mm, 5 μm |
|
Mobile phase |
Acetonitrile : phosphate/formate buffer pH ~3.0 (20:80, v/v) |
|
Flow rate |
1.0 mL/min (range 0.8–1.2 mL/min) |
|
Column temperature |
25–30 °C |
|
Detection wavelength |
276–277 nm (UV/PDA) |
|
Injection volume |
10–20 μL |
|
Retention time (prucalopride) |
~5–6 min |
|
Total run time |
~10 min |
|
Theoretical plates |
> 2000 |
|
Tailing factor |
≤ 1.5 |
Table 1. Representative optimized RP-HPLC conditions reported for prucalopride.
4. METHOD VALIDATION:
Once an optimized chromatographic condition has been selected from within the Method Operable Design Region, the method must be formally validated in accordance with ICH Q2(R1), and, where the more recent framework is adopted, the harmonized ICH Q2(R2)/Q14 guideline on analytical procedure development and validation 35,36. Validation demonstrates, with documented evidence, that the method is suitable for its intended purpose. The following subsections describe each validation parameter as applied to a stability-indicating RP-HPLC method for prucalopride, drawing on the approach and acceptance criteria consistently reported across the pharmaceutical analytical literature for structurally comparable basic drug substances 9,27,28,29.
4.1 Specificity
Specificity (selectivity) is demonstrated by injecting blank diluent, placebo (tablet excipient blend without active), standard prucalopride solution, and forced-degradation samples generated under each stress condition, and confirming that the prucalopride peak is fully resolved from the diluent/placebo response and from every degradation product peak, with no co-elution. Peak-purity assessment using a PDA detector comparing spectra collected at the peak up-slope, apex and down-slope, and confirming a purity angle less than the purity threshold provides additional, spectroscopic confirmation that the prucalopride peak is homogeneous and free from a co-eluting degradant, which is the analytical basis on which a method's "stability-indicating" claim ultimately rests.
4.2 Linearity
Linearity is established by preparing and analysing a minimum of five to six concentration levels spanning an appropriate range around the target assay concentration typically 50-150% of the nominal working concentration for a stability-indicating assay method, or a wider range (for example, 25-150 μg/mL or equivalent) if the method is also intended to quantify low-level degradation products. A calibration curve of peak area (y-axis) versus concentration (x-axis) is constructed, and linear regression analysis is performed; a correlation coefficient (r²) of ≥ 0.999 is generally required to demonstrate satisfactory linearity, a criterion consistently met by reported prucalopride RP-HPLC methods 9,32.
4.3 Accuracy
Accuracy is evaluated through recovery studies, in which known quantities of prucalopride reference standard are spiked into a pre-analysed sample matrix (commonly tablet powder blend) at three concentration levels typically 50%, 100% and 150% of the target assay concentration each prepared and analysed in triplicate. Percentage recovery is calculated as (amount found/amount added) × 100, with an acceptance range of 98-102% (or 97-103%, depending on the level and internal specification) considered indicative of satisfactory accuracy; comparable stability-indicating RP-HPLC methods for structurally related basic drug substances have reported recoveries in the range of approximately 98.6-103.4% 9,10,31.
4.4 Precision
Precision is assessed at two levels. Repeatability (intra-day precision) is determined by analysing six replicate preparations of the same sample concentration on the same day, under the same operating conditions, and calculating the percentage relative standard deviation (%RSD) of the assay results; an %RSD ≤ 2% is the generally accepted criterion. Intermediate precision (ruggedness) extends this evaluation across different days, different analysts, and/or different instruments within the same laboratory, again with an acceptance criterion of %RSD ≤ 2%. Together, repeatability and intermediate precision confirm that the method generates consistent results under both routine and varied operating conditions.
4.5 Limit of Detection and Limit of Quantification
The limit of detection (LOD) and limit of quantification (LOQ) are determined either from the standard deviation of the response and the slope of the calibration curve (LOD = 3.3σ/S; LOQ = 10σ/S) or by signal-to-noise ratio approaches (typically 3:1 for LOD and 10:1 for LOQ). Reported LOD/LOQ values for prucalopride by RP-HPLC and UV-spectrophotometric methods fall in the sub-microgram to low-microgram-per-millilitre range, for example LOD/LOQ values of approximately 0.01/0.03 μg/mL and 0.014/0.047 μg/mL reported under two different UV-spectrophotometric conditions, indicating that RP-HPLC and related chromatographic techniques provide more than adequate sensitivity for assay and stability applications, and are further capable, with appropriate optimization, of quantifying trace-level degradation products and impurities 14.
4.6 Robustness
Robustness is evaluated by deliberately introducing small, systematic variations to method parameters identified as critical during the QbD risk-assessment exercise typically mobile-phase pH (± 0.2 units), organic-modifier ratio (± 2-5%), flow rate (± 0.1 mL/min), and column temperature (± 5 °C) and confirming that system suitability parameters (resolution, tailing factor, theoretical plates) and assay results remain within acceptance limits despite these deliberate perturbations. A key advantage of AQbD-derived methods is that robustness testing essentially re-confirms, at the extremes of the previously established Method Operable Design Region, a property that was already demonstrated computationally during the DoE optimization step, giving greater confidence that the method will perform reliably during routine use and inter-laboratory transfer.
4.7 System Suitability
System suitability testing is performed prior to and interspersed throughout each analytical run to verify that the chromatographic system is performing adequately at the time of use. Typical acceptance criteria applied to prucalopride RP-HPLC methods include: theoretical plate count > 2000; tailing (asymmetry) factor ≤ 1.5-2.0; percentage RSD of peak area and retention time for six replicate injections of the standard solution ≤ 2%; and, where a stability-indicating claim is made, resolution ≥ 2.0 between the prucalopride peak and the nearest-eluting degradation product 5,9.
4.8 Solution Stability
Solution stability (sample and standard solution stability on the bench-top and, where relevant, under refrigeration) is evaluated by preparing standard and sample solutions and re-analysing them at defined time intervals (for example, initial, 12, 24 and 48 hours) against a freshly prepared reference, confirming that no significant degradation (typically < 2% change in assay value) occurs over the period during which solutions are expected to be used in the laboratory. This parameter ensures that observed differences in assay results reflect genuine sample variability rather than analyte instability in solution.
|
Validation Parameter |
ICH Requirement / Acceptance Criterion |
Reported for Prucalopride |
|
Specificity |
Resolution from all degradants; spectral purity (PDA) |
Demonstrated under 6 stress conditions |
|
Linearity |
r² ≥ 0.999; ≥5 concentration levels |
50–150% of working concentration; r² ≥ 0.999 |
|
Accuracy |
Recovery 98–102% (or 97–103%) |
~98.6–103.4% across 50/100/150% levels |
|
Repeatability |
%RSD ≤ 2%; n = 6 |
Reported ≤ 2% |
|
Intermediate precision |
%RSD ≤ 2%; different days/analysts |
Reported ≤ 2% |
|
LOD |
S/N ≥ 3:1 or 3.3σ/S |
Sub-μg/mL range |
|
LOQ |
S/N ≥ 10:1 or 10σ/S |
Sub-μg/mL range |
|
Robustness |
SST within limits at ±pH 0.2, ±organic 2–5%, ±flow 0.1 mL/min |
Confirmed within MODR boundaries |
|
System suitability |
Plates > 2000; tailing ≤ 1.5–2.0; %RSD ≤ 2% |
Met in all reported methods |
|
Solution stability |
< 2% change over expected use period |
Evaluated at 12, 24, 48 h |
Table 2. Summary of ICH Q2(R1)/Q2(R2) validation parameters for prucalopride RP-HPLC methods.
5. FORCED DEGRADATION STUDIES:
5.1 Rationale and ICH Q1A(R2) Framework
Stress (forced) degradation testing is explicitly required by ICH Q1A(R2) as part of new drug substance and drug product development, with the stated purpose of establishing the inherent stability characteristics of the molecule, identifying likely degradation products and pathways, and critically for this review validating the stability-indicating power of the analytical procedure intended for use in formal stability studies 33. A method that has never been challenged with genuinely degraded material cannot be assumed to separate the intact drug from every degradation product that might arise over a two- or three-year shelf life; forced degradation, conducted under conditions considerably harsher than any real-world storage condition, provides that assurance in an accelerated timeframe.
5.2 Objectives of Forced Degradation for Prucalopride
The specific objectives of a forced degradation study for prucalopride are to: (i) generate an adequate but not excessive degree of degradation (generally targeted at 5-20% loss of parent drug, avoiding both insufficient stress and complete destruction of the analyte) under each stress condition; (ii) confirm mass balance, i.e., that the sum of the residual parent drug and the quantified degradation products accounts for close to 100% of the initial drug content, thereby indicating that no degradation product has escaped detection; (iii) demonstrate that the prucalopride peak remains chromatographically resolved and spectrally pure in the presence of all generated degradation products; and (iv) where feasible, characterise and tentatively identify the major degradation products and propose a plausible degradation pathway.
5.3 Stress Conditions
5.3.1 Acid Hydrolysis
Acid hydrolysis is performed by treating a stock solution of prucalopride with dilute hydrochloric acid (typically 0.1-1 N HCl), at room temperature or with mild heating, for a defined period, followed by neutralisation prior to injection. Reported studies indicate that prucalopride is susceptible to measurable degradation under acidic conditions, consistent with hydrolytic cleavage of the amide bond linking the benzofuran-carboxamide core to the piperidinyl side-chain, a structural feature common to benzamide/carboxamide-based enterokinetic agents 9,10.
5.3.2 Alkaline Hydrolysis
Base (alkaline) hydrolysis is carried out using dilute sodium hydroxide (typically 0.1-1 N NaOH) under conditions analogous to the acid stress study, again followed by neutralisation. Amide-containing molecules such as prucalopride are frequently more susceptible to base-catalysed hydrolysis than to acid hydrolysis, and the literature on prucalopride forced degradation consistently reports alkaline conditions among the more aggressive degradation pathways, generating one or more resolvable degradation product peaks distinct in retention time from the parent drug 9,10.
5.3.3 Oxidative Degradation
Oxidative degradation is induced using hydrogen peroxide (typically 3-30% v/v H2O2) at room temperature or with mild heating for a defined exposure period. Given the presence of an aromatic primary amine and a tertiary piperidine nitrogen in the prucalopride structure, both of which are susceptible to oxidation to N-oxide or related oxidative products, oxidative stress is generally reported to produce measurable degradation, with degradation products eluting separately from the parent peak; N-oxide formation has, in fact, been specifically identified as a characterised degradation-related impurity of prucalopride in the literature.
5.3.4 Thermal (Dry-Heat) Degradation
Thermal (dry heat) degradation is performed by exposing the solid drug substance or its solution to elevated temperature (commonly 60-105 °C) for an extended period (typically 24-72 hours), simulating accelerated and stress storage conditions. Prucalopride has generally been reported to show relatively modest degradation under dry-heat stress alone compared with hydrolytic and oxidative conditions, consistent with the reasonable thermal stability implied by its melting point of approximately 198 °C, though some reduction in assay and minor peak formation is typically still observed.
5.3.5 Photolytic Degradation
Photolytic degradation is assessed in accordance with ICH Q1B, exposing the drug substance (as solid and/or in solution) to defined exposure of UV and visible/fluorescent light (typically not less than 1.2 million lux-hours of visible light and 200 watt-hours/m² of UV light, or equivalent), alongside a suitably protected (dark) control. This condition is included specifically because certain aromatic amine- and benzofuran-containing structures are known to be photolabile, and confirms whether special packaging or storage precautions (light-protective packaging) are warranted for prucalopride drug product.
5.3.6 Neutral Hydrolysis
Neutral hydrolysis is evaluated by refluxing or heating an aqueous solution of prucalopride (without added acid, base or oxidant) for an extended period, providing a control condition that helps distinguish degradation attributable specifically to hydrolytic catalysis by H+ or OH- ions from degradation that would occur through simple aqueous exposure and heat alone.
5.4 Overall Stability Profile and Susceptibility Ranking
Across the reported literature, prucalopride has generally been found to be most labile under alkaline and oxidative stress conditions, moderately labile under acidic hydrolysis, and comparatively stable under dry-heat, neutral-hydrolysis and photolytic conditions, although the precise rank order and percentage degradation reported varies somewhat between studies depending on the specific stress intensity (acid/base normality, peroxide concentration, exposure duration and temperature) employed 9,10. This overall susceptibility pattern is consistent with the presence of a hydrolytically labile amide linkage and oxidisable nitrogen centres within the prucalopride structure, and underscores the importance of a genuinely comprehensive, ICH-aligned stress panel rather than a restricted subset of conditions when establishing the stability-indicating credentials of a new method.
5.5 Peak Purity Assessment
Peak purity assessment using a photodiode-array detector is the principal tool used to confirm that the prucalopride peak observed in each stressed sample chromatogram is spectrally homogeneous that is, that the UV spectrum collected across the peak's up-slope, apex and down-slope regions is invariant, and that the calculated purity angle remains below the purity threshold angle generated by the instrument software. A purity angle exceeding the threshold angle for any stressed sample would indicate co-elution of a degradation product beneath the parent peak and would necessitate further method refinement (typically adjustment of the organic modifier ratio or gradient profile) before the method could be considered genuinely stability-indicating.
5.6 Mass Spectrometric Characterisation of Degradation Products
Where mass spectrometric characterisation has been undertaken for example using LC-QTOF-MS/MS in conjunction with RP-HPLC separation major degradation products of prucalopride generated under acidic and alkaline stress have been tentatively attributed to hydrolytic cleavage products arising from amide bond scission, while oxidative stress has been associated with N-oxidation of the piperidine or aromatic amine nitrogen. Such hyphenated LC-MS characterisation, going beyond simple UV/PDA-based specificity demonstration, provides structural confidence in the proposed degradation pathway and can support impurity qualification and toxicological risk assessment of the identified degradants.
5.7 Proposed Degradation Pathway
Based on the collective hydrolytic and oxidative degradation behaviour reported for prucalopride, a plausible overall degradation pathway can be proposed in which the parent molecule undergoes (i) acid- and base-catalysed hydrolytic cleavage of the carboxamide bond linking the dihydrobenzofuran ring system to the 1-(3-methoxypropyl)piperidin-4-yl moiety, yielding the corresponding carboxylic acid and amine fragments, and (ii) oxidation at the tertiary piperidine nitrogen and/or the aromatic primary amine to the corresponding N-oxide derivative under peroxide stress. This proposed pathway remains broadly consistent with the general degradation chemistry expected of amide-containing, amine-substituted heteroaromatic drug substances, though full structural confirmation for each degradation product would require dedicated LC-MS/NMR characterisation beyond the scope of the routine stability-indicating assay method itself.
5.8 Basis for Stability-Indicating Validation
Taken together, the resolution of the prucalopride peak from all degradation products generated across the acid, base, oxidative, thermal, photolytic and neutral stress panels, combined with satisfactory peak-purity (spectral homogeneity) results and acceptable mass balance, is the basis on which reported RP-HPLC methods for prucalopride have been validated as genuinely stability-indicating, fulfilling the specificity requirement of ICH Q2(R1)/Q2(R2) and the stress-testing expectation of ICH Q1A(R2) simultaneously 9,10,33,35.
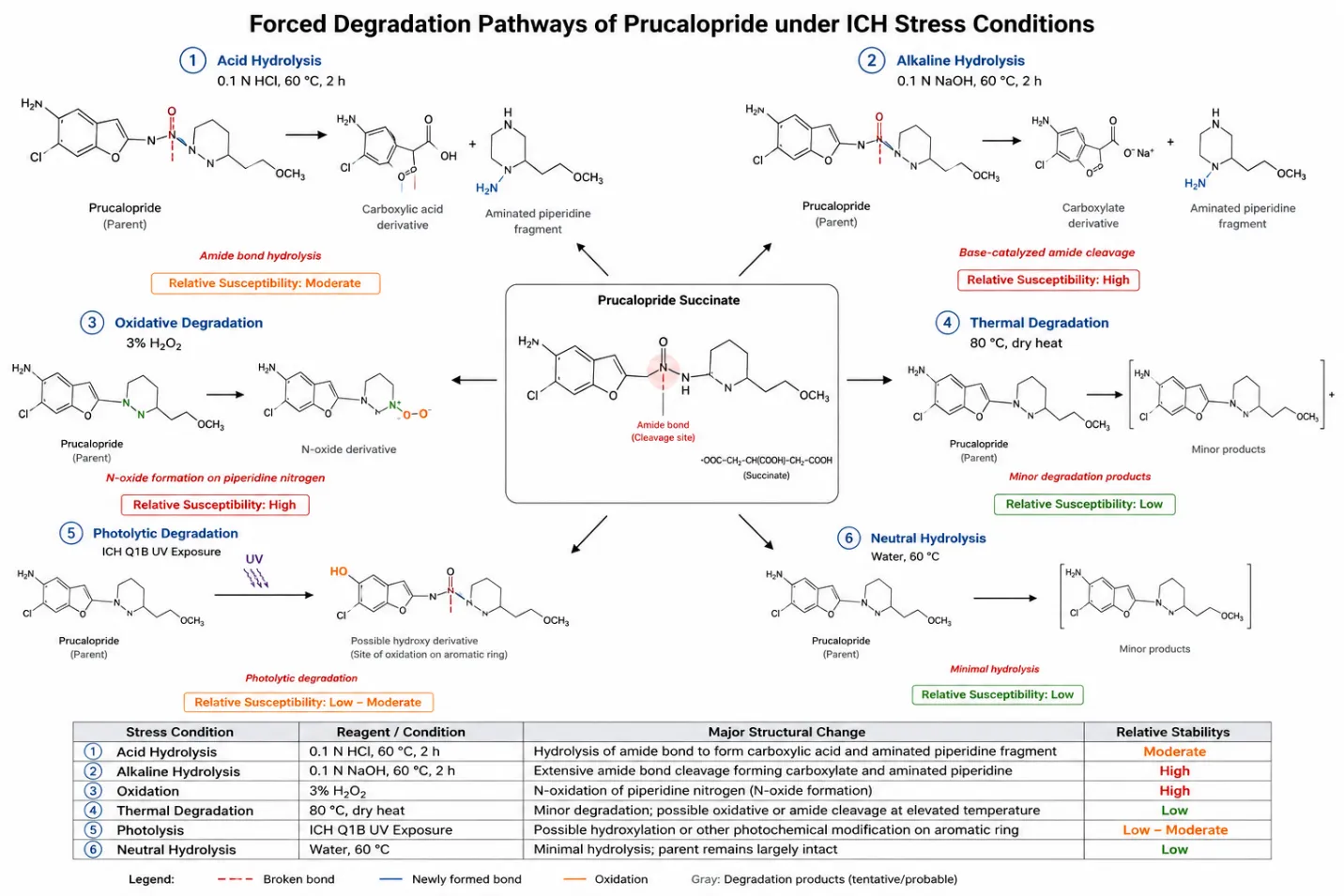
Figure 2: Proposed Degradation Pathways of Prucalopride under ICH Q1A(R2) Stress Conditions
|
Stress Condition |
Reagent / Condition |
Reported Susceptibility |
Key Degradation Products |
|
Acid hydrolysis |
0.1–1 N HCl, RT or mild heat |
Moderate |
Amide hydrolysis products |
|
Alkaline hydrolysis |
0.1–1 N NaOH, RT or mild heat |
High |
Carboxylic acid + amine fragments |
|
Oxidative |
3–30% Hâ‚‚Oâ‚‚, RT |
High |
N-oxide derivatives |
|
Thermal (dry heat) |
60–105 °C, 24–72 h |
Low–Moderate |
Minor uncharacterised peaks |
|
Photolytic |
ICH Q1B (≥1.2 × 10ⶠlux·h visible) |
Low–Moderate |
Aromatic amine photo-products |
|
Neutral hydrolysis |
Water, elevated temperature |
Low |
Minor peaks |
Table 3. Summary of forced degradation behaviour of prucalopride under ICH Q1A(R2)-aligned stress conditions.
6. COMPARATIVE EVALUATION OF ANALYTICAL METHODS FOR PRUCALOPRIDE:
A range of analytical techniques has been reported in the literature for the quantification of prucalopride, spanning UV-spectrophotometric, spectrofluorimetric, electrochemical, RP-HPLC/UPLC and LC-MS/MS approaches, each suited to different applications (bulk/formulation quality control versus biological-matrix pharmacokinetic or bioequivalence studies). Table 3 summarises the principal reported methods, and the discussion below critically compares their relative strengths and limitations, with particular emphasis on the advantages offered by a QbD-optimized, stability-indicating RP-HPLC method over these alternatives.
6.1 Discussion of Reported Analytical Techniques
UV-spectrophotometric methods for prucalopride, whether by direct absorbance measurement or by derivative spectroscopy, offer simplicity, low cost and wide accessibility, and are entirely adequate for simple assay of the bulk drug or of a single-component tablet formulation where no significant excipient or degradant interference is expected 14,31. However, UV methods lack the selectivity required to resolve prucalopride from its degradation products or from co-formulated actives, and are therefore fundamentally unsuited to stability-indicating applications; a UV method can, at best, be used as a rapid screening tool alongside a validated chromatographic method, never as a substitute for one. Spectrofluorimetric and electrochemical (potentiometric/voltammetric) methods offer improved sensitivity and, in some cases, notable "green chemistry" credentials, but similarly lack the chromatographic separation needed to distinguish parent drug from degradation products and are generally reported for bioanalytical or environmental-monitoring applications rather than for pharmaceutical stability testing. LC-MS/MS and UHPLC-MS/MS methods, by contrast, offer outstanding sensitivity (reported linear ranges as low as 50 pg/mL to low ng/mL in plasma) and are the method of choice for pharmacokinetic, bioavailability and bioequivalence studies where prucalopride must be quantified in complex biological matrices at trace concentrations 11,12; however, the capital cost, technical complexity and specialised expertise associated with mass spectrometric instrumentation make LC-MS/MS impractical as a routine, high-throughput quality-control tool in a typical pharmaceutical manufacturing or contract-testing laboratory.
6.2 Advantages of QbD-Optimized RP-HPLC over Alternative Methods
Against this backdrop, a QbD-optimized RP-HPLC method with UV/PDA detection occupies a distinctive and practically important niche: it delivers the chromatographic selectivity necessary for a genuinely stability-indicating assay something UV-spectrophotometric, fluorimetric and electrochemical methods cannot offer while remaining accessible to routine quality-control laboratories using conventional, widely available HPLC instrumentation, unlike LC-MS/MS. The QbD/DoE-based development process further ensures that this selectivity and separation are robust across the normal operating range of the method, reducing the risk of method failure, out-of-specification results, or costly re-development after initial validation and technology transfer.
|
Technique |
Matrix |
Linear Range |
LOD / LOQ |
Stability-Indicating? |
Key Advantage |
|
UV Spectrophotometry |
Bulk / tablet |
~5–50 μg/mL |
~0.01/0.03 μg/mL |
No |
Simple; low cost |
|
Derivative UV |
Bulk / tablet |
Comparable |
Low μg/mL |
No |
Reduces baseline noise |
|
Spectrofluorimetry |
Bulk / biological |
Sub-μg/mL |
Very low |
No |
High sensitivity; green |
|
Electrochemical |
Bulk / environmental |
Variable |
ng/mL range |
No |
High sensitivity |
|
RP-HPLC (OFAT) |
Bulk / tablet |
50–150% LC |
Sub-μg/mL |
Yes* |
Selective; routine use |
|
RP-HPLC (AQbD) |
Bulk / tablet |
50–150% LC |
Sub-μg/mL |
Yes |
Robust; MODR-defined |
|
UPLC |
Bulk / tablet |
Comparable |
Comparable |
Yes |
Shorter run time |
|
LC-MS/MS |
Plasma / biological |
pg/mL – ng/mL |
pg/mL range |
Partially |
Bioanalytical; trace |
* Requires validated forced degradation challenge to confirm specificity.
Table 4. Comparative summary of analytical methods reported for prucalopride.
7. APPLICATIONS ACROSS THE PHARMACEUTICAL PRODUCT LIFECYCLE:
A validated, stability-indicating, QbD-optimized RP-HPLC method for prucalopride has direct application across several stages of the pharmaceutical product life cycle. In routine assay of marketed tablet formulations, the method provides accurate, precise quantification of label-claim potency for batch release and ongoing quality monitoring. In content-uniformity testing, the same chromatographic conditions, applied to individual dosage units, confirm consistency of active ingredient distribution across a batch, a particularly important consideration for the low-dose (1 mg and 2 mg) prucalopride tablets where content-uniformity risk is inherently higher than for higher-dose products. In dissolution testing, the method (potentially adapted with a shorter run time for high sample throughput) can quantify prucalopride released into dissolution medium at each sampling time-point, supporting in vitro performance characterisation and biowaiver justification. Most significantly, the stability-indicating capability of the method makes it directly applicable to formal ICH-conforming long-term, intermediate and accelerated stability studies, where the method must reliably detect and quantify any degradation products that form during real-time or accelerated storage, supporting shelf-life assignment and, where necessary, root-cause investigation of stability failures. Collectively, these applications underscore the central role that a single, well-developed analytical method plays across quality control, regulatory submission and post-approval life-cycle management.
8. ADVANTAGES AND LIMITATIONS OF THE AQbD APPROACH:
The principal advantages of a QbD-based, stability-indicating RP-HPLC method for prucalopride include: demonstrable robustness, since the method's tolerance to small variations in critical parameters has already been established computationally across the design space before formal robustness testing is even performed; a documented, statistically justified rationale for every chromatographic condition selected, which strengthens regulatory dossiers and simplifies responses to agency queries; reduced overall development time and material consumption relative to sequential OFAT optimization, since DoE extracts more information from fewer experimental runs; and a Method Operable Design Region that provides built-in flexibility for minor post-validation adjustments without necessarily triggering full re-validation. Balanced against these advantages are certain limitations: AQbD requires access to, and working knowledge of, statistical design and response-surface-modelling software, together with a level of statistical literacy not universally available in every analytical laboratory; the upfront investment of time in risk assessment and screening/optimization experiments, although smaller than the cumulative cost of an OFAT approach, is still non-trivial and may appear a barrier for very simple, low-risk methods; and the quality of the final design space is only as good as the risk assessment that defined which factors were studied in the first place, meaning that an incomplete or poorly reasoned risk assessment can produce a false sense of security about method robustness.
9. REGULATORY CONSIDERATIONS:
Prucalopride does not, at the time of writing, carry an individual monograph in the major pharmacopoeias (United States Pharmacopeia, British Pharmacopoeia or European Pharmacopoeia), meaning that method developers must rely on regulatory guidance documents rather than a compendial fixed method. The principal applicable ICH guidelines are Q1A(R2) for stress/stability testing design 33, Q1B for photostability testing 34, Q2(R1) and the newer harmonized Q2(R2)/Q14 for analytical procedure validation and development respectively 35,36, and Q8(R2), Q9 and Q10 for the broader pharmaceutical-development, quality-risk-management and pharmaceutical-quality-system principles from which the AQbD philosophy is drawn 37,38,39. In the absence of a compendial method, a well-documented, in-house-developed and independently validated stability-indicating RP-HPLC method, supported by a transparent AQbD development rationale, becomes the primary evidentiary basis on which a marketing-authorisation applicant or manufacturer must rely when submitting analytical data to a regulatory authority such as the US FDA, EMA, Health Canada or an equivalent national agency.
10. FUTURE DIRECTIONS:
Several directions are likely to shape future analytical work on prucalopride and structurally similar molecules. Green analytical chemistry principles are increasingly being incorporated into method development, with reported prucalopride methods already exploring reduced-solvent, eco-scale-assessed spectrofluorimetric and electrochemical alternatives; extending green-chemistry metrics (such as the Analytical Eco-Scale or Complex-GAPI index) to RP-HPLC method optimization, for example by substituting acetonitrile with less hazardous organic modifiers, represents a natural next step. Chemometric and machine-learning-assisted approaches to DoE analysis, capable of modelling more complex, higher-order interactions than conventional quadratic response-surface models, offer the potential for further-refined design spaces with fewer experimental runs. Finally, wider application of hyphenated LC-MS/MS and LC-QTOF-MS/MS techniques for structural characterisation of prucalopride degradation products would strengthen the currently only partially characterised degradation pathway, supporting more complete impurity qualification and, ultimately, a future compendial monograph for the drug substance.
CONCLUSION
Prucalopride succinate, as a first-in-class 5-HT4 receptor agonist without an existing compendial monograph, presents an instructive and practically important case for the application of Quality by Design principles to RP-HPLC method development. Systematic risk assessment, DoE-based screening and optimization (typically Box-Behnken or central composite designs applied to mobile-phase pH, organic modifier ratio and flow rate), and definition of a Method Operable Design Region together provide a scientifically defensible, efficient pathway to a robust chromatographic method one demonstrably more resilient to routine operational variability than a method arrived at through traditional one-factor-at-a-time optimization. When this QbD-optimized method is further challenged and validated against a comprehensive ICH Q1A(R2)-aligned forced degradation panel (acid, base, oxidative, thermal, photolytic and neutral hydrolysis), and shown by peak-purity and mass-balance analysis to resolve prucalopride cleanly from all degradation products, the resulting method meets the full definition of a validated, stability-indicating assay under ICH Q2(R1)/Q2(R2). Such a method is directly applicable to routine assay, content uniformity, dissolution and most importantly formal stability studies of prucalopride drug substance and drug product, and represents a considerably stronger regulatory and quality-assurance asset than UV-spectrophotometric or other non-chromatographic alternatives, while remaining more practical for routine quality-control use than specialised LC-MS/MS bioanalytical platforms. This review consolidates the currently available literature on both fronts AQbD-based RP-HPLC method development and prucalopride-specific forced degradation behaviour into a single reference framework intended to guide future method-development and validation work on this and structurally related molecules.
REFERENCES

Nidhi Uday Kalamkar*, Pravin P. Gadak, Nikhil A. Bawane, Pallavi D. Borse, Mamta D. Wadkar, Analytical Method Development And Validation Of Prucalopride By Quality By Design (Qbd) Approach And Stability-Indicating Forced Degradation Study Using RP-HPLC, Int. J. Sci. R. Tech., 2026, 3 (7), 553-569. https://doi.org/10.5281/zenodo.21425044
 10.5281/zenodo.21425044
10.5281/zenodo.21425044