
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmaceutical Quality Assurance, S.S.P. Shikhan Sanstha's Siddhi College of Pharmacy, Chikhali, Pune, Maharashtra - 411062
Process validation is an important requirement of Good Manufacturing Practices (GMP) that helps ensure pharmaceutical products are consistently manufactured with the desired quality, safety, and effectiveness. Over the years, the concept and application of process validation have developed significantly, leading to the adoption of the lifecycle approach. This approach focuses on understanding and controlling the manufacturing process from product development through commercial production and continuous monitoring. The present review explains the basic principles of life cycle process validation in a simple manner and high lights its key stages. It also demonstrates how these principles can be applied in pharmaceutical manufacturing using the example of a sterile inject able product. Proper implementation of lifecycle process validation helps maintain product quality and supports compliance with regulatory requirements worldwide.
​Until the 1970s, the pharmaceutical industry relied heavily on testing finished products to ensure quality. However, serious outbreaks caused by contamination with E. cloacae and Erwinia in large volume parenteral (LVP) bottles led to fatalities and major societal losses. Surprisingly, these bottles were found to be contaminated even after sterilization.
The root cause was not the failure of sterilization itself, but oversight in quality practices. During the cooling phase after sterilization, moisture entered the space beneath the screw cap, allowing microorganisms to contaminate the product. This contamination went undetected in routine quality tests because the cap and septum were removed before testing, masking the issue.1
The development of a pharmaceutical product is a complex and time-consuming process that includes drug discovery, laboratory experimentation, animal testing, clinical studies, and regulatory approval. Process control activities involve inspection of raw materials, in-process monitoring, and evaluation of the final product against established specifications. The main objective is to assess and monitor both online and offline performance of the manufacturing process and subsequently validate it.
Even after successful validation of the manufacturing process, current Good Manufacturing Practices (cGMP) require the establishment of well-documented procedures for process control to ensure continuous monitoring and consistent performance of the process.
Process validation is primarily guided by the regulations of the U.S. Food and Drug Administration regarding current Good Manufacturing Practices for finished pharmaceutical products, which are outlined in 21 CFR Parts 210 and 211. These regulations state that manufacturing processes must be properly designed and controlled to ensure that both in-process materials and finished products consistently meet predetermined quality standards and specifications. Therefore, process validation is considered an essential requirement under cGMP regulations described in Parts 210 and 211.
This incident exposed significant gaps in the industry’s quality assurance approach. It highlighted that relying solely on end-product testing was insufficient. As a result, the industry recognized the need to ensure that manufacturing processes themselves are reliable and capable. This led to the concept of process qualification or validation—demonstrating and documenting that a process can consistently produce products meeting the desired quality standards.1
The validation protocol is examined and authorized by the Head of Quality Assurance, Head of Quality Engineering, Validation Manager, Production Manager, and experts from the validation department. It serves as a documented plan that describes the procedure for evaluating the manufacturing process and product quality against predefined acceptance criteria to ensure the quality, safety, and efficacy of the product.
The validation protocol generally consists of the following elements:
1. Objectives and scope of the validation process.
2. Category or type of validation to be conducted.
3. Number of batches selected for validation studies.
4. Validation team members, including their qualifications and assigned responsibilities.
5. Critical process parameters and quality attributes along with specified limits.
6. Statistical techniques used for interpretation and analysis of data.
7. Approved analytical methods for testing raw materials and in-process samples with established specifications.
8. Calibration and qualification requirements for equipment and manufacturing processes.
9. Proper recording of observations, conclusions, and authorization of validation study outcomes.
HISTORY OF VALIDATION
The concept of process validation was initially introduced in the mid-1970s by two FDA officials, Ted Byers and Bud Loftus, with the goal of enhancing pharmaceutical quality (Agalloco, 1995). This initiative was prompted by recurring issues related to the sterility of large-volume parenteral products. Early validation efforts primarily targeted the manufacturing processes of these products but soon expanded to include other related pharmaceutical processes.
Although the U.S. FDA was a pioneer in promoting process validation, a formal definition of the term did not appear in any FDA publications or regulations until September 29, 1978. Prior to this date, current Good Manufacturing Practice (cGMP) regulations did not specifically address process validation 5
EVOLUTION OF CONCEPT OF PROCESS VALIDATION
When the pharmaceutical industry first encountered the term “validation” through GMP guidelines, its expectations and practical applications were not well defined. In 1987, the regulatory landscape advanced significantly when the FDA issued the first formal guidance on process validation in the life sciences field. This guidance introduced a transformative idea: quality cannot simply be tested into a finished product; instead, it must be built into the product from the beginning.2,3
Process validation was defined as the establishment of documented evidence providing a high level of assurance that a specific process will consistently produce a product that meets predetermined specifications and quality attributes. The guideline emphasized the importance of implementing controls at every stage of the manufacturing process, even at the smallest steps.4,11
The purpose of process validation was to demonstrate that a process is both effective and reproducible, thereby reducing reliance on in-process and final product testing. However, it was also recognized that end-product testing remains necessary. Together, process validation and final product testing provide greater confidence that the finished product meets the required quality standards.
The 1987 US FDA 12,13guidance established the basic principles of process validation, but it did not clearly convey its full requirements or significance. At that time, process validation was largely treated as a documentation exercise, typically involving the collection of extensive data from three consecutive validation batches. Despite this effort, it did not always ensure the expected level of product quality, and regulatory inspections continued to highlight deficiencies due to lack of robust, validated processes.
Most regulatory authorities, including the United States Food and Drug Administration, (12,13)European Medicines Agency13,14Health Canada15, and Indian16,regulatory bodies, have adopted this approach and issued regional guidelines to align with the current recommendations of the Pharmaceutical Inspection Co-operation Scheme17,18International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use, and the World Health Organization19, Additionally, both regulatory agencies and the pharmaceutical industry are working toward compliance with ISO 9000 standards, which focus on quality management systems (QMS). These standards support organizations in fulfilling customer expectations while ensuring adherence to statutory and regulatory requirements for products and services.20,21
Regulatory authorities worldwide—including the U.S. Food and Drug Administration, European Medicines Agency, Health Canada, and national regulators in India—have adopted this lifecycle approach. Their guidelines align with international standards from organizations such as the Pharmaceutical Inspection Co-operation Scheme and the World Health Organization.
Additionally, both industry and regulators emphasize compliance with ISO 9000 standards, which focus on quality management systems designed to meet customer and regulatory requirements. As expectations continue to evolve, pharmaceutical companies must continuously update their manufacturing and validation practices to stay aligned with current global standards.
OBJECTIVE OF PROCESS VALIDATION
Process Validation (PV) is a Quality Assurance (QA) activity that confirms the quality of a drug product by generating documented evidence that the manufacturing process consistently performs as intended.
Validation ensures effective control over the process, demonstrating consistent performance and reliable repeatability. Generally, three consecutive successful production batches are considered sufficient to establish process consistency and reproducibility.5
DEFINITION OF PROCESS VALIDATION
The concept of validation, particularly process validation, has developed significantly over time. According to the 2011 guidance “Process Validation: General Principles and Practices” issued by the U.S. Food and Drug Administration, process validation is defined as the collection and analysis of data from the process design stage through to commercial production, providing scientific evidence that a process can consistently produce a quality product.
In the modern context, validation is no longer limited to pre-marketing activities. Instead, it is an on going effort that spans the entire product lifecycle. This approach involves a series of structured activities divided into three stages, each contributing to building confidence in the reliability of the manufacturing process and the consistency of the product’s quality attributes13
DEFINITIONS
The concept of validation has been defined by various regulatory authorities with a common focus on ensuring consistent quality outcomes.
The European Commission describes validation as the act of demonstrating, in compliance with GMP, that a process reliably produces the expected results. It further emphasizes that validation involves documented evidence showing that a process, when operated within established parameters, performs effectively and consistently to produce a medicinal product meeting predefined specifications and quality attributes.26
According to the U.S. Food and Drug Administration, process validation is the establishment of documented evidence that provides a high level of assurance that a specific process will consistently yield a product that meets its predetermined specifications and quality standards.27
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use defines process validation as a method of ensuring and documenting that processes, when operated within defined design parameters, are capable of repeatedly and reliably producing a finished product of the desired quality.29
Similarly, the World Health Organization defines validation as the documented process of proving that any procedure, equipment, material, activity, or system consistently leads to the intended results.30
NEED FOR PROCESS VALIDATION
REASONS FOR PROCESS VALIDATION
Process validation is carried out for several important reasons, such as the introduction of a new product or modifications made to an existing product. It is also required when changes occur in manufacturing facilities, batch size, critical process parameters (CPPs), or when there is a change in the supplier of the Active Pharmaceutical Ingredient (API) or critical excipients. In addition, process validation becomes necessary when recurring Out of Specification (OOS) results or trends are observed among production batches. Overall, process validation serves as an effective approach for establishing, maintaining, and assuring product quality and process consistency.26
Challenges Associated with Process Validation
In the pharmaceutical industry, process validation is an essential activity to ensure product quality, consistency, and regulatory compliance. However, several common mistakes can weaken the effectiveness of validation programs.
Understanding these errors and applying suitable corrective measures is important for meeting the requirements of regulatory authorities such as the FDA, EMA, and MHRA.
Understanding Process Validation
Process validation is the documented evidence proving that a manufacturing process consistently produces products that meet predetermined quality standards and specifications. Regulatory agencies recommend a lifecycle approach to validation, which includes Process Design, Process Qualification, and Continued Process Verification. Recognizing and correcting common errors during these stages is necessary to maintain product quality and compliance.
Poor Risk Assessment
One major mistake in process validation is inadequate risk assessment. Failure to properly identify potential risks can negatively affect product quality and process reliability.
To avoid this issue:
Proper risk assessment helps organizations identify possible failures and implement effective control measures.
Inadequate Sampling
Insufficient or unrepresentative sampling can produce inaccurate validation results and incorrect conclusions regarding process capability.
To improve sampling practices:
Effective sampling ensures that validation data accurately reflects the overall manufacturing process.
Weak Validation Protocols
Poorly written validation protocols may result in incomplete studies, inconsistent execution, and regulatory non-compliance. Validation protocols should clearly describe objectives, methods, acceptance criteria, and expected outcomes.
To strengthen protocols:
Well-structured protocols improve consistency and reliability in validation activities.
Missing or Incomplete Data
Missing data can create uncertainty about process performance and product quality. This problem mayoccur due to poor documentation, human errors, or incomplete sampling.
To prevent missing data:
Accurate data management strengthens the credibility of validation outcomes.
Continuous Improvement and Feedback
Continuous improvement is essential for maintaining an effective validation program. Organizations should learn from previous validation experiences and use the information to improve future practices.
Important steps include:
A continuous improvement approach helps organizations maintain compliance, improve process efficiency, and build a strong quality culture.
Product Lifecycle Approach
Validation is a key component of GMP and an integral part of the quality management system. Its primary purpose is to ensure that a process can consistently produce a product that meets the desired quality standards. Unlike the traditional approach, which relied heavily on testing and documentation, the modern concept of validation adopts a scientific, lifecycle-based perspective.
This updated approach connects all stages of a product’s lifecycle—ranging from process and product development to validation of the commercial manufacturing process, and ongoing regulatory maintenance to keep the process under control during routine production. The concept emerged from a vision established during a 2003 ICH meeting in Brussels, which aimed to create a harmonized pharmaceutical quality system applicable across the entire product lifecycle, with a strong focus on science and quality risk management.
Guidelines such as International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use Q8, Q9, and Q10 outline this modern, science- and risk-based approach. They promote the use of tools like Quality by Design (QbD) and Quality Risk Management (QRM), integrated within a robust Pharmaceutical Quality System (PQS). QbD, in particular, is defined as a systematic development approach that begins with predefined objectives and emphasizes a deep understanding of both the product and the process, supported by sound science and risk management principles. The U.S. Food and Drug Administration has also issued guidance to encourage the adoption of QbD in the industry.
By combining comprehensive pharmaceutical development with risk management principles (as described in ICH Q9), this approach focuses on gaining in-depth knowledge of the product and manufacturing process while identifying potential sources of variability. Controlling these variations enhances product quality assurance and reduces reliance on end-product testing. Early understanding also helps optimize process parameters to accommodate variability in raw materials.
Furthermore, the Pharmaceutical Quality System described in ICH Q10 supports continuous improvement, leading to better process understanding and performance over time. Altogether, this integrated, science- and risk-based framework strengthens quality control strategies, improves confidence in product quality, and provides greater flexibility in manufacturing operation
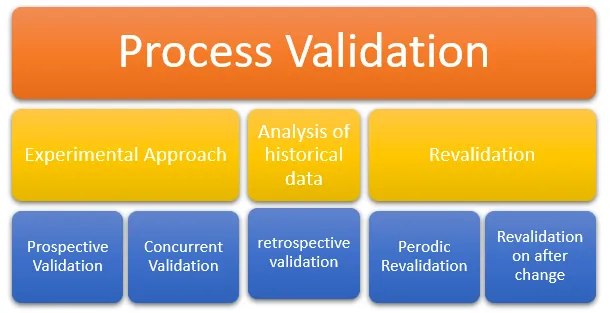
Types of Process Validation
Process validation is classified into different types depending on the stage at which the validation study is performed in relation to the manufacturing process. The main types are as follows:
Prospective Validation
Prospective validation is performed before a product is distributed for the first time or whenever major modifications are introduced in the manufacturing process that may influence product quality and characteristics. It is based on risk assessment and prior knowledge to identify possible critical situations that could affect the process.6The risks and their causes are carefully evaluated, and trial protocols are developed according to the required priorities. Trial batches are then manufactured, monitored, and assessed. If the results are satisfactory, the process is approved. If not, necessary improvements are made until the process consistently meets validation requirements. This type of validation helps minimize risks before commercial production begins.
Concurrent Validation
Concurrent validation is carried out during routine production, where products manufactured during validation batches are released for sale after successful qualification.7 This approach is suitable only when the manufacturing process is already well understood through development studies. Usually, the first three commercial-scale batches are monitored extensively. It involves continuous monitoring of Critical Process Parameters (CPPs) and testing of finished products to demonstrate that the process remains under control. Proper justification and authorized approval are required before conducting concurrent validation.
Retrospective Validation
Retrospective validation involves evaluating historical production data and past manufacturing experience to confirm process consistency. This method assumes that the formulation, equipment, procedures, and operating conditions have remained unchanged over time. It is mainly applied to products already available in the market and should not be used for new products or processes. 9The batches selected for review must represent both successful and unsuccessful batches to provide a complete assessment of process performance. The primary aim of retrospective validation is to establish confidence in the reliability and consistency of the manufacturing process.5
Revalidation
Revalidation is required whenever significant changes are made to critical process parameters, formulation, equipment, manufacturing procedures, or packaging materials. Its purpose is to ensure that these modifications do not negatively affect product quality or process performance. Revalidation may also be necessary when a process fails to meet established specifications. In addition, facilities, systems, equipment, and processes should be reviewed periodically to confirm that they continue to operate in a validated state. If no major changes have occurred, documented evidence demonstrating continued compliance with predefined requirements may be sufficient to satisfy revalidation requirements.
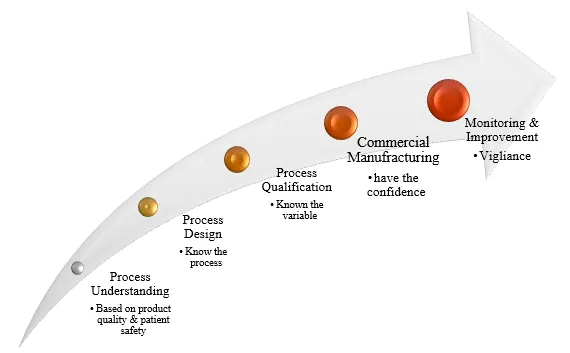
Stages of Process Validation
Under the modern approach, process validation is structured into three distinct stages. These stages, along with their respective sub-stages, are typically illustrated in Figure 1.
|
Country/ region |
Regulator |
Applicable GMP regulations |
|
India |
State Drugs Control Administration (DCA) and Central Drugs Standard Control Organization (CDSCO) |
WHO GMP and Indian GMP that is, Schedule M (cGMP and Requirements of Premises, Plant and Equipment for Pharmaceutical Products) and Schedule U (Manufacturing Records) of Drugs and Cosmetics Act 1940 regulate the manufacture and sale of pharmaceuticals in India.22 |
|
European Union (EU) |
European Medicines Agency or EMA (coordinating/harmonizing role at EU level; coordinating GMP inspections of manufacturing sites for medicines whose marketing authorization is submitted through the centralized procedure or as part of a referral procedure). National competent authority of 31 EEA countries are responsible for inspecting manufacturing sites responsible for manufacture/import within their own territories. |
Regulation No. 1252/2014 and Directive 2003/94/EC, apply to active substances and medicines for human use and Directive 91/412/ EEC applies to medicines for veterinary use. In addition, Directive 2001/83/EC and Directive 2001/82/EC lay down related provisions. Further, Regulation No. 2017/745 is applicable for Medical Devices.25 |
|
US |
US Food & Drug Administration or USFDA |
21 CFR Part 210 (Current Good Manufacturing Practice in Manufacturing Processing, packing, or Holding of Drugs) and 211 Regulations (Current Good Manufacturing Practice for Finished Pharmaceuticals) and 21 CFR Part 820 (for Medical Devices) set the current GMP requirements.23,24 |
|
Canada |
Health Canada or HC |
Part C, Division 2 of the Food and Drug Regulations C.02.001 to C.02.030 regulate activities of fabricators, packagers, labellers, testers, distributors, importers and wholesalers of drugs in Canada31 |
|
Australia |
Therapeutic Goods Administration or TGA |
Section 36 of the Therapeutic Goods Act 1989 regulate Medicinal products supplied in Australia and requires that manufacturers meet the PIC/S Guide to Good Manufacturing Practice (GMP) - 01 May 2021, PE009-15 issued by Pharmaceutical Inspection Co-operation Scheme, except for its Annexes 4, 5 and 14 which are not adopted by Australia.25 The Australian Code of Good Manufacturing Practice (GMP) for Blood and Blood Components, Human Tissues and Human Cellular Therapy Products (the Code) applies to Blood, Human Tissues and Human Cellular Therapy Products manufacturers that undertake the collection, processing, testing, storage, release for supply, and quality assurance of Human Blood and Blood Components, Human Tissues and Human Cellular Therapy Products.26 |
|
China |
The National Medical Products Administration (NMPA), formerly the China Food and Drugs Administration |
Good Manufacturing Practice for Drugs (2010 Revision), MOH Decree No. 79 in accordance with the Drug Administration Law of the People’s Republic of China and the Regulations for Implementation of the Drug Administration Law of the People’s Republic of China, regulate the manufacturing and quality management of Drugs in China.3 |
|
Brazil |
AgênciaNacional de VigilânciaSanitária - ANVISA |
Following ANVISA regulations are applicable: Drug Products (Medicines) - Resolution RDC 658/2022 (general aspects) and Normative Instructions associated. Radiopharmaceuticals - Resolution RDC 658/2022 and Normative Instruction - IN 128/2022. Medicinal Gases - Resolution RDC 658/2022 and Normative Instruction - IN 129/2022. Herbal products - Resolution RDC 658/2022 and Normative Instruction - IN 130/2022. Active Pharmaceutical Ingredients (APIs) -Resolution RDC 654/2022 (GMP) and RDC 204/2006 (Good Distribution and Fractionating Practices for Pharmaceutical Supplies). Pharmaceutical Excipients - Resolution RDC 34/2015. Medical Devices - Resolution RDC 665/2022 (general aspects); Resolution RDC 687/2022 (administrative processes).32 |
|
Japan |
Pharmaceutical and Medical Devices Agency or PMDA |
“Ministerial Ordinance on Standards for The “Ministerial Ordinance on Standards for Manufacturing Control and Quality Control for Drugs and Quasi-drugs” (MHLW Ministerial Ordinance No. 179, 2004) establishes the GMP requirements for the manufacturing and quality control of drugs and quasi-drugs in Japan. Furthermore, the PIC/S Guide to Good Manufacturing Practice for Medicinal Products provides additional guidance for facilities engaged in the manufacture and quality control of investigational medicinal products, in accordance with Paragraph 1, Article 17 and Article 26-3 of the GMP regulations for investigational medicinal products under MHLW Ministerial Ordinance No. 28, 1997. |
Table 1: Major regional regulatory bodies and applicable GMP regulations
Stage 1 – Process Design
Stage 2 – Process Qualification
Stage 3 – Continuous Process Verification
Stage 1 – Process Design
The objective of this stage is to develop a commercial manufacturing process capable of consistently producing a product that meets the desired quality attributes. It includes all activities related to product development, such as research and development, selection of the active pharmaceutical ingredient (API) and excipients, choice of dosage form, formulation development, process development, pilot-scale studies, and technology transfer to commercial-scale production. The knowledge gained during this phase supports the creation of a comprehensive master validation plan.
Process Design Components
Knowledge Management (KM)
relies on a strong foundation of product and process knowledge, which is obtained from two main sources:
Prior knowledge serves as the starting point for designing the commercial process. This may include published literature, past experience with similar products or processes, laboratory research, preclinical and clinical study data An effective process design, analytical characterization, and an understanding of the physicochemical and biological properties of the drug substance, as well as compatibility data.
Pharmaceutical development studies build on this foundation by generating additional knowledge through experiments conducted during the development of the commercial-scale process.
Science- and Risk-Base Approach
The combined knowledge from prior experience and development studies provides a scientific basis for designing a robust manufacturing process at a commercial scale. When this scientific understanding is integrated with risk-based principles, it ensures a more reliable and well-controlled process.
Risk Assessment in Process Validation
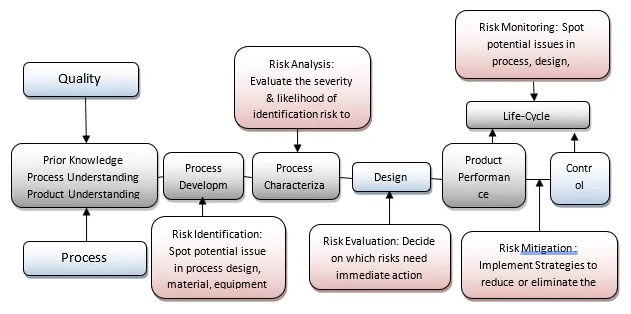
Process Validation

REFERENCES


Priya Vaware*, Priyanka Nimbalkar, Muskan Shaikh, Hitanshi Darji, P. N. Sable, Lifecycle Approach To Process Validation And Its Implementation In Pharmaceutical Industry, Int. J. Sci. R. Tech., 2026, 3 (6), 1643-1654. https://doi.org/10.5281/zenodo.21032467
 10.5281/zenodo.21032467
10.5281/zenodo.21032467